Diagnosis
Marfan syndrome can be challenging for doctors to diagnose because many connective tissue disorders have similar signs and symptoms. Even among members of the same family, the signs and symptoms of Marfan syndrome vary widely — both in their features and in their severity.
Certain combinations of symptoms and family history must be present to confirm a diagnosis of Marfan syndrome. In some cases, a person may have some features of Marfan syndrome, but not enough of them to be diagnosed with the disorder.
Heart tests
If your doctor suspects Marfan syndrome, one of the first tests he or she may recommend is an echocardiogram. This test uses sound waves to capture real-time images of your heart in motion. It checks the condition of your heart valves and the size of your aorta. Other heart-imaging options include computerized tomography (CT) scans and magnetic resonance imaging (MRI).
If you are diagnosed with Marfan syndrome, you'll need to have regular imaging tests to monitor the size and condition of your aorta.
Eye tests
Eye exams that may be needed include:
- Slit-lamp exam. This test checks for lens dislocation, cataracts or a detached retina. Your eyes will need to be completely dilated with drops for this exam.
- Eye pressure test. To check for glaucoma, your eye doctor may measure the pressure inside your eyeball by touching it with a special tool. Numbing eyedrops are usually used before this test.
Genetic testing
Genetic testing is often used to confirm the diagnosis of Marfan syndrome. If a Marfan mutation is found, family members can be tested to see if they are also affected. You may want to talk to a genetic counselor before starting a family, to see what your chances are of passing on Marfan syndrome to your future children.
More Information
Treatment
While there is no cure for Marfan syndrome, treatment focuses on preventing the various complications of the disease. To accomplish this, you'll need to be checked regularly for signs that the damage caused by the disease is progressing.
In the past, people who had Marfan syndrome often died young. With regular monitoring and modern treatment, most people with Marfan syndrome can now expect to live a more normal life span.
Medications
Doctors often prescribe blood pressure lowering drugs to help prevent the aorta from enlarging and to reduce the risk of dissection and rupture.
Therapy
The vision problems associated with a dislocated lens in your eye often can be corrected with glasses or contact lenses.
Surgical and other procedures
Ascending aortic root aneurysm repair and replacement
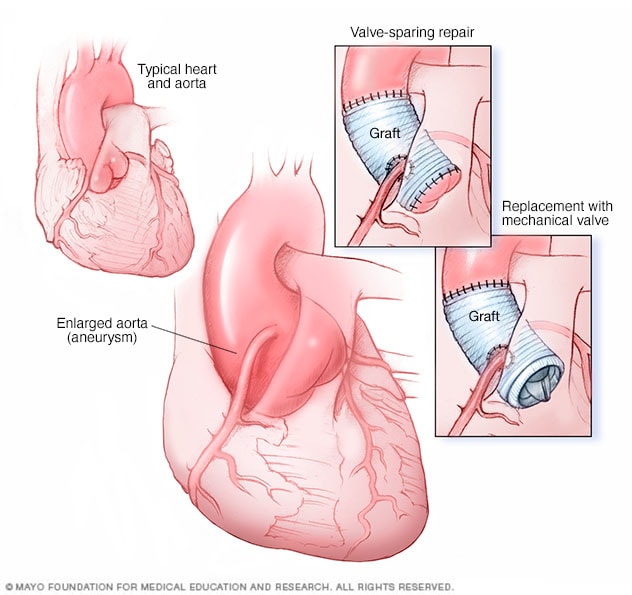
Ascending aortic root aneurysm repair and replacement
Ascending aortic root aneurysm repair and replacement may be done in two ways. Valve-sparing aortic root repair (top-right image) replaces the enlarged part of the aorta with an artificial tube, called a graft. The aortic valve stays in place. In aortic valve and aortic root replacement (bottom-right image), the valve and a part of the aorta are removed. A graft replaces the part of the aorta. A mechanical or biological valve replaces the valve.
Depending on your signs and symptoms, procedures might include:
- Aortic repair. If your aorta's diameter reaches about 2 inches (50 millimeters) or if it enlarges rapidly, your doctor may recommend an operation to replace a portion of your aorta with a tube made of synthetic material. This can help prevent a life-threatening rupture. Your aortic valve may need to be replaced as well.
- Scoliosis treatment. When there is significant scoliosis, a consultation with a spine expert is necessary. Bracing and surgery are needed in some cases.
- Breastbone corrections. Surgical options are available to correct the appearance of a sunken or protruding breastbone. Because these operations are often considered to be for cosmetic purposes, your insurance might not cover the costs.
- Eye surgeries. If parts of your retina have torn or come loose from the back of your eye, surgical repair is usually successful. If you have cataracts, your clouded lens can be replaced with an artificial lens.
Video: Valve-sparing aortic root replacement
Alberto Pochettino, M.D., Cardiovascular Surgery, Mayo Clinic: My name is Alberto Pochettino. I'm a cardiovascular surgeon at Mayo Clinic in Rochester, Minnesota. I have a special interest in aortic surgery, and today I will discuss the operation known as valve-sparing aortic root replacement.
The aortic root is the beginning of the aorta. It is located at the transition between the main pumping chamber of the heart and the remainder of the aorta. It contains the aortic valve and the origin of the coronary arteries. Replacement of the aortic root is indicated in the presence of an aortic aneurysm. An aneurysm is an abnormal enlargement of a blood vessel. The maximum diameter of the aneurysm is used to assess the risk of rupture or dissection. At the level of the aortic root, 5.5 centimeter in maximum diameter is felt to be the size at which replacement surgery should be performed in most patients. Conditions in which the aortic wall is intrinsically weaker mandate intervention at a lower size. Most of these conditions are due to genetic abnormality of the aortic wall. The classic abnormality is Marfan syndrome but other rare abnormalities have been defined such as Ehler-Danlos syndrome, Loeys-Dietz syndrome and a few others. However, by far the most common genetic aortic abnormality affecting the aortic root is bicuspid aortic valve disease. In all of these patients with high risk conditions, the trigger for surgical intervention should be lowered to 5 centimeters for many bicuspid aortic valve and 4.5 centimeters for most Marfan and other more severe genetic abnormalities.
Historically, replacement of the aortic root required replacement of the aortic valve contained within it, even when the valve may not have been significantly diseased. In a younger individual, a mechanical aortic valve would have been recommended because of its durability, but it requires life-long anticoagulation with blood thinners such as Coumadin. The alternative tissue aortic valve would not require Coumadin but has limited lifespan, necessitating re-operation. The reluctance to replace the aortic root when found within a normal aortic valve prompted development in the 1980s of techniques to spare the native valve. The first attempt came to be known as remodeling, followed by the reimplantation technique first reported by Tirone David. The reimplantation technique over subsequent years has proved to be the most durable. The surgical principle is to replace the entire aortic root from the ventricular annular junction to the so-called sinutubular junction with a Dacron graft tube. Within this tube, the three-dimensional aortic valve is reimplanted, thus the name. Over the subsequent years, the aortic root reimplantation, sometimes known as David operation, has been shown to be safe and effective in a specialized center performed by dedicated aortic surgeons. While long-term results have been very positive, in some patients the native aortic valve may still deteriorate, requiring long-term monitoring and in some individuals, eventual replacement.
Over the years, factors have been identified that impact the successful outcome of the procedure. For example, the larger the aneurysm, the more distortion the aortic valve leaflet will suffer, leading to worse aortic valve insufficiency. The more longstanding the aortic insufficiency, the more fibrosis and abnormality occur in the leaflets, leading to a lower success in sparing the valve and a lower long-term durability of the spared valve. This has led to earlier intervention in select patients to allow for a durable valve function within the replaced root.
Despite this general tendency of earlier surgery, my practice has continued to use maximum aortic diameter combined with genetic risk factors to justify root replacement. Early in the development of valve-sparing root replacement, bicuspid aortic valves were not considered eligible due to the intrinsic abnormality of those valves. More recently, good results have been achieved in the treatment of root aneurysm where bicuspid aortic valves are well functioning. While long-term outcomes in this set of patients may be less optimal than in the trileaflet aortic valve, a spared bicuspid valve is likely to be quite durable as well. Unfortunately, a significant fraction of bicuspid valves in patients with root aneurysms are not normal and still require valve replacement.
In summary, aortic root replacement should be dictated by the size of the aortic root aneurysm as well as the genetic risk factors. When aortic valve leaflets are of good quality and can be spared, they should be. However, if the leaflets have significant abnormalities and the repair is not likely to be durable, the valve should be replaced with an appropriate prosthesis to meet the needs of the individual patient. Clinical judgment should be used both when deciding when to operate -- such that premature intervention is avoided -- as well as at surgery -- to maximize the benefits to the patient, even if that may occasionally mean replacement of the aortic valve.
More Information
Lifestyle and home remedies
You may need to avoid competitive sports and certain recreational activities if you're at increased risk of aortic dissection or rupture. Increases in blood pressure, common in activities such as weightlifting, place extra strain on the aorta. Less intense activities — such as brisk walking, bowling, doubles tennis or golf — are generally safer.
Coping and support
Living with a genetic disorder can be extremely difficult for both adults and children. Adults may wonder how the disease will affect their careers, their relationships and their sense of themselves. And they may worry about passing the defective gene to their children.
But Marfan syndrome can be even harder on young people, especially because the often-inherent self-consciousness of childhood and adolescence may be exacerbated by the disease's effect on appearance, academic performance and motor skills.
Helping children cope
Working together, parents, teachers and medical professionals can provide children with both emotional support and practical solutions for some of the more distressing aspects of the disease. For example, children with Marfan syndrome may struggle in school because of vision problems that can be corrected with glasses or contact lenses.
For most young people, cosmetic concerns are at least as important as academic ones. Parents can help by anticipating these concerns and offering solutions, such as:
- Contact lenses instead of glasses
- A brace for scoliosis
- Dental work for crowded teeth
- Clothes that flatter a tall, thin frame
Support groups
People who have Marfan syndrome often find it helpful to talk with others facing the similar challenges. The Marfan Foundation provides a variety of support services online.
Preparing for your appointment
Marfan syndrome can affect many different parts of your body, so you may need to see a variety of medical specialists, such as:
- A cardiologist, a doctor who specializes in heart and blood vessel disorders
- An ophthalmologist, a doctor who specializes in eye disorders
- An orthopedist, a doctor who specializes in structural problems of the skeleton
- A geneticist, a doctor who specializes in genetic disorders
To make the best use of appointment time, plan ahead and have important information available, including:
- Detailed descriptions of all your symptoms
- Details of your past medical history, including any previous surgeries
- Past X-rays and echocardiogram reports, which often can be sent electronically
- A list of all your medications and supplements
What to expect from your doctor
All your doctors will want to hear about your specific symptoms, and whether anyone in your family has had Marfan syndrome or experienced an early, unexplained heart-related disability or death.