Sept. 12, 2018
The interstitial pneumonias (IPs) are a heterogeneous group of diffuse parenchymal lung diseases characterized by specific clinical, radiologic and pathologic features. While pathologically defined, significant overlap in terms of presentation as well as association with secondary diseases is known and may confound initial work-up and diagnosis. This review focuses on recent changes and additions to definitions and diagnostic criteria with implications for management.
Revised interstitial pneumonia classification
Characterizing features of interstitial pneumonias
A newly revised classification system includes eight pathologically defined interstitial pneumonias.
Eight pathologically defined interstitial pneumonias are included in a newly revised classification system, published in the American Journal of Respiratory and Critical Care Medicine in 2013.
- Usual interstitial pneumonia (UIP)
- Nonspecific interstitial pneumonia (NSIP)
- Cryptogenic organizing pneumonia (COP)
- Desquamative interstitial pneumonia (DIP)
- Respiratory bronchiolitis-interstitial lung disease (RB-ILD)
- Acute interstitial pneumonia (AIP)
- Lymphoid interstitial pneumonia (LIP)
- Idiopathic pleuroparenchymal fibroelastosis (PPFE)
Suspected PPFE in female with progressive dyspnea and hypoxemia
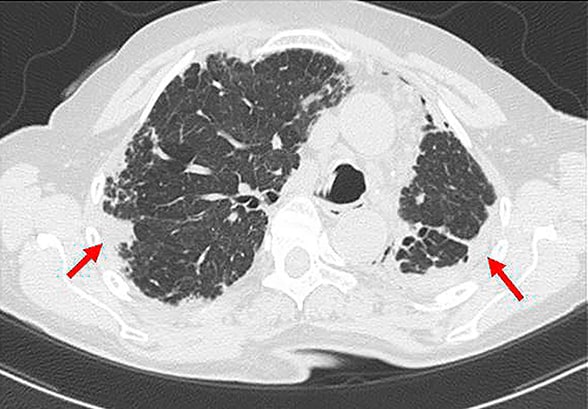
Suspected PPFE in female with progressive dyspnea and hypoxemia
Suspected PPFE in a 73-year-old female with progressive dyspnea and hypoxemia. Note upper-lobe-predominant pleural thickening with significant volume loss of the left lung and compensating hyperinflation of the right lung (red arrows). Underlying interstitial pneumonia pattern appeared consistent with possible UIP characterized by bibasilar reticular and mild honeycomb changes.
An additional category, "unclassifiable," has also been added to include interstitial pneumonia not fitting a particular pathologic pattern. PPFE, the newest pathologic subcategory, is rare and highlighted by pleural thickening predominantly in the upper lobes. It is often associated with parenchymal or interstitial findings on CT, most commonly UIP and NSIP. Given the rarity of presenting cases, a confident diagnosis of PPFE is likely best achieved by biopsy as clinical and radiologic presentation alone may be equivocal.
While IPs have been studied and recognized over several decades, the new classification system provides a more intuitive organization of both the prevalence and natural course of specific histologic patterns and their related clinical findings. A first approach is to separate the eight pathologically defined patterns into six major (UIP, NSIP, COP, DIP, RB-ILD, AIP) and two rare or less commonly encountered entities (LIP and PPFE).
Of the six major patterns, a review of their courses and presentations as well as associated clinical findings further leads to three subcategorizations:
- Chronically fibrosing (UIP and NSIP)
- Smoking related (DIP and RB-ILD)
- Acutely presenting (COP and AIP)
This approach may better assist the clinician in terms of recognition and work-up of initially undifferentiated presenting disease. While any of the eight may appear independently as primary or idiopathic disease, many are involved in the progressive lung injury associated with chronic organic or inorganic exposures, drug toxicity, and autoimmune disease. A combined approach of not only characterizing the presenting clinical and radiologic features but also seeking a secondary cause is important to diagnosis and subsequent management.
Interstitial pneumonia with autoimmune features (IPAF)
Prior studies have suggested differences in survival and clinical course for interstitial lung disease (ILD) with specifically elicited clinical and serologic features of autoimmune disease. While several definitions have been previously proposed, a recent international consensus statement, published in American Journal of Respiratory and Critical Care Medicine in 2013, has delineated specific criteria for interstitial pneumonias with incompletely diagnosed but suggestive autoimmune disease, currently described as interstitial pneumonia with autoimmune features (IPAF).
Exact criteria involve the confirmation of an interstitial process by radiologic or pathologic presentation, exclusion of other associated causes including defined connective tissue disease and at least two features from three representative clinical domains. These domains include specific autoimmune clinical signs and symptoms, positive findings on any of 12 autoimmune serologies, and morphologic findings of interstitial pneumonia. Remaining morphologic criteria also include nonparenchymal and extrapulmonary features such as evidence of serositis with pleural or pericardial disease, vasculopathy, or intrinsic airway disease.
Acute exacerbation in male presenting with ILD fitting IPAF criteria
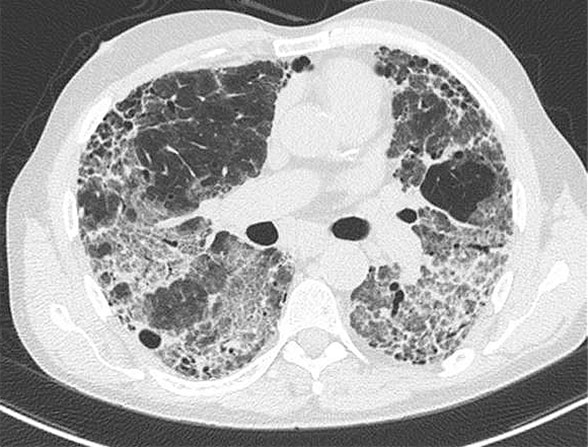
Acute exacerbation in male presenting with ILD fitting IPAF criteria
Acute exacerbation in a 57-year-old male presenting with ILD fitting IPAF criteria (positive antinuclear antibodies titer > 1:2560, Raynaud's phenomenon and possible UIP CT pattern). Rapid decline over several weeks was noted while on immunosuppressive therapy, where patient presented profoundly hypoxemic and was ultimately diagnosed with Pneumocystis jiroveci pneumonia. This case highlights two important discussion points: 1. The inclusion of UIP in IPAF criteria where UIP findings on CT appear to progress in a similar fashion to idiopathic pulmonary fibrosis. 2. While Pneumocystis jiroveci pneumonia was eventually diagnosed, new definitions would frame this under the category of a triggered acute exacerbation and not simply infectious pneumonia.
It is important to note the inclusion of UIP pathology and radiologic patterns despite prior studies assessing the presence of autoimmune serology or clinical symptoms in these patients, noting little difference in their clinical course or survival as compared to those with idiopathic pulmonary fibrosis.
Questions remain as to the utility of these disease criteria in clinical practice and implications for long-term management or follow-up. It is unknown how many initial IPAF evolve to diagnosable connective tissues over time, and if connective tissue disease is not diagnosed, whether survival is simply reflective of the underlying histopathology where UIP often portends poorer outcome as compared with NSIP or other histologic patterns.
Reframing acute exacerbation
Acute exacerbation (AE) represents punctuated decline in respiratory function (less than 30 days) with new and superimposed infiltrates in the setting of idiopathic pulmonary fibrosis. The exclusion of secondary causes, including performance of bronchoscopy or tracheal aspirate to assess infection, is key to diagnosis.
Despite well-defined criteria, a standardized approach to initial work-up remains elusive as institutional approaches vary and complete exclusion of secondary causes is often difficult in real-world practice. An example is the reluctance associated with performing bronchoscopy in patients who are not intubated and presenting with significant respiratory distress and hypoxemia. This aversion is not unfounded as further decompensation leading to intubation and mechanical ventilation is known to be associated with greater morbidity and mortality in this setting.
On the other hand, delay in performing bronchoscopy — and the selection of obtained microbiologic studies — may theoretically decrease its yield, particularly when broad-spectrum antibiotics are often empirically provided. The term "suspected acute exacerbation" was therefore recently advocated for acute worsening of respiratory symptoms unexplained by secondary causes but with incomplete work-up.
Recent updates to the international consensus definition of AE, published in the American Journal of Respiratory and Critical Care Medicine in 2016, have reflected on these difficulties and modified prior criteria in the hopes of better reflecting clinical practice and outcomes. New definitions no longer require complete exclusion of secondary causes, but instead include known findings as triggers of AE. An initial approach is to ensure the absence of pulmonary edema or volume overload where AE may be excluded, followed by a reasonable assessment for secondary etiologies where known and unspecified causes of respiratory failure are all categorized as forms of AE. This approach contrasts with the prior definition, where exclusion of secondary causes was important to diagnosis, in effect framing AE as an idiopathic phenomenon.
In many ways, the discussion correlates with the Berlin definition of acute respiratory distress syndrome (ARDS), a conceptual model where severity of hypoxemia along with bilateral infiltrates and clinical absence of heart failure frame the acute event. Indeed, associated triggers such as pneumonia, aspiration, septicemia or pancreatitis in acute respiratory distress syndrome are part and parcel of the work-up and management, but the focus is directed at broadly managing the acute respiratory failure syndrome, which may behave independently of the original inciting etiology.
Whether this model holds similar implications for the future management of acute exacerbation in ILD is yet unknown, as historical use of low tidal volume strategies has not proved beneficial. In fact, mechanical ventilation appears to be associated with worse survival, though it is unknown whether mechanical ventilation truly causes additional harm in this setting or is simply a surrogate of more-severe and perhaps irreversible lung injury.
For more information
Travis WD, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. American Journal of Respiratory and Critical Care Medicine. 2013; 188:733.
Collard HR, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. American Journal of Respiratory and Critical Care Medicine. 2016: 194;265.