Overview
Phenylketonuria (fen-ul-key-toe-NU-ree-uh), also called PKU, is a rare inherited disorder that causes an amino acid called phenylalanine to build up in the body. PKU is caused by a change in the phenylalanine hydroxylase (PAH) gene. This gene helps create the enzyme needed to break down phenylalanine.
Without the enzyme necessary to break down phenylalanine, a dangerous buildup can develop when a person with PKU eats foods that contain protein or eats aspartame, an artificial sweetener. This can eventually lead to serious health problems.
For the rest of their lives, people with PKU — babies, children and adults — need to follow a diet that limits phenylalanine, which is found mostly in foods that contain protein. Newer medications may allow some people with PKU to eat a diet that has a higher or an unrestricted amount of phenylalanine.
Babies in the United States and many other countries are screened for PKU soon after birth. Although there is no cure for PKU, recognizing PKU and starting treatment right away can help prevent limitations in areas of thinking, understanding and communicating (intellectual disability) and major health problems.
Products & Services
Symptoms
Newborns with PKU initially don't have any symptoms. However, without treatment, babies usually develop signs of PKU within a few months.
Signs and symptoms of untreated PKU can be mild or severe and may include:
- A musty odor in the breath, skin or urine, caused by too much phenylalanine in the body
- Nervous system (neurological) problems that may include seizures
- Skin rashes, such as eczema
- Lighter skin, hair and eye color than family members, because phenylalanine can't transform into melanin — the pigment responsible for hair and skin tone
- Unusually small head size (microcephaly)
- Hyperactivity
- Intellectual disability
- Delayed development
- Behavioral, emotional and social problems
- Mental health disorders
Severity varies
The severity of PKU depends on the type.
- Classic PKU. The most severe form of the disorder is called classic PKU. The enzyme needed to break down phenylalanine is missing or severely reduced. This results in high levels of phenylalanine that can cause severe brain damage.
- Less severe forms of PKU. In mild or moderate forms, the enzyme still has some function, so phenylalanine levels are not as high, resulting in a smaller risk of significant brain damage.
Regardless of the form, most infants, children and adults with the disorder still require a special PKU diet to prevent intellectual disability and other complications.
Pregnancy and PKU
Women who have PKU and become pregnant are at risk of another form of the condition called maternal PKU. If women don't follow the special PKU diet before and during pregnancy, blood phenylalanine levels can become high and harm the developing baby.
Even women with less severe forms of PKU may place their unborn children at risk by not following the PKU diet.
Babies born to women with high phenylalanine levels don't often inherit PKU. But a child can have serious problems if the level of phenylalanine is high in the mother's blood during pregnancy. At birth, the baby may have:
- Low birth weight
- Unusually small head
- Problems with the heart
In addition, maternal PKU can cause the child to have delayed development, intellectual disability and problems with behavior.
When to see a doctor
Talk to your health care provider in these situations:
- Newborns. If routine newborn screening tests show that your baby may have PKU, your child's health care provider will want to start dietary treatment right away to prevent long-term problems.
- Women of childbearing years. It's especially important for women with a history of PKU to see a health care provider and maintain the PKU diet before becoming pregnant and during pregnancy. This reduces the risk of high blood phenylalanine levels harming their unborn babies.
- Adults. People with PKU need to receive lifelong care. Adults with PKU who have stopped the PKU diet in their teens may benefit from a visit with their health care providers. Returning to the diet may improve mental functioning and behavior and prevent further damage to the central nervous system that can result from high phenylalanine levels.
Causes
Autosomal recessive inheritance pattern
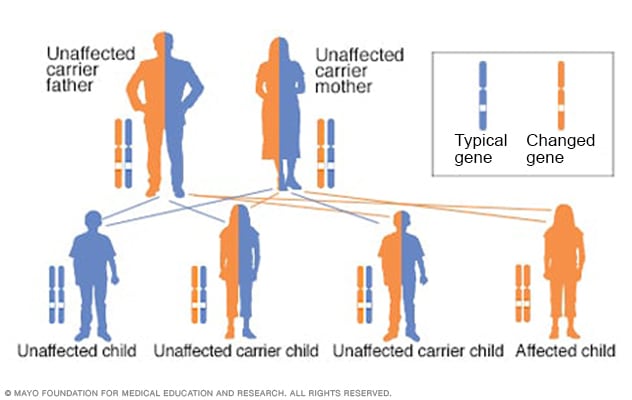
Autosomal recessive inheritance pattern
To have an autosomal recessive disorder, you inherit two changed genes, sometimes called mutations. You get one from each parent. Their health is rarely affected because they have only one changed gene. Two carriers have a 25% chance of having an unaffected child with two unaffected genes. They have a 50% chance of having an unaffected child who also is a carrier. They have a 25% chance of having an affected child with two changed genes.
A gene change (genetic mutation) causes PKU, which can be mild, moderate or severe. In a person with PKU, a change in the phenylalanine hydroxylase (PAH) gene causes a lack of or reduced amount of the enzyme that's needed to process phenylalanine, an amino acid.
A dangerous buildup of phenylalanine can develop when a person with PKU eats protein-rich foods, such as milk, cheese, nuts or meat, or grains such as bread and pasta, or aspartame, an artificial sweetener.
Inheritance
For a child to inherit PKU, both the mother and father must have and pass on the changed gene. This pattern of inheritance is called autosomal recessive.
It's possible for a parent to be a carrier — to have the changed gene that causes PKU, but not have the disease. If only one parent has the changed gene, there's no risk of passing PKU to a child, but it's possible for the child to be a carrier.
Most often, PKU is passed to children by two parents who are both carriers of the changed gene, but don't know it.
Risk factors
Risk factors for inheriting PKU include:
- Having both parents with a gene change that causes PKU. Two parents must pass along a copy of the changed gene for their child to develop the condition.
- Being of a certain racial or ethnic descent. PKU affects people from most ethnic backgrounds worldwide. But in the United States, it's most common in people of European ancestry and much less common in people of African ancestry.
Complications
Untreated PKU can lead to complications in infants, children and adults with the disorder. When women with PKU have high blood phenylalanine levels during pregnancy, it can harm their unborn baby.
Untreated PKU can lead to:
- Irreversible brain damage and marked intellectual disability beginning within the first few months of life
- Neurological problems such as seizures and tremors
- Behavioral, emotional and social problems in older children and adults
- Major health and developmental problems
Prevention
If you have PKU and are considering getting pregnant:
- Follow a low-phenylalanine diet. Women with PKU can prevent harm to their developing baby by sticking to or returning to a low-phenylalanine diet before becoming pregnant. Nutritional supplements designed for people with PKU can ensure enough protein and nutrition during pregnancy. If you have PKU, talk to your health care provider before you start trying to conceive.
- Consider genetic counseling. If you have PKU, a close relative with PKU or a child with PKU, you may benefit from genetic counseling before becoming pregnant. A specialist in medical genetics (geneticist) can help you better understand how PKU is passed through your family. The specialist can also help determine your risk of having a child with PKU and assist with family planning.