April 16, 2021
A 39-year-old man presented for evaluation of recurrent fractures associated with extensive fibrous dysplasia of bone. His initial fracture occurred at the tibia at age 10 with minimal trauma. During childhood he sustained multiple additional fractures, including:
- His right humerus on four occasions
- A traumatic right hip fracture in grade school requiring treatment with an intramedullary rod and interlocking screws
The patient's puberty started at age 12. Following high school, he noted a gradual increase in facial asymmetry with prominence of his forehead and left check. He denied prior chronic glucocorticoid or anti-epileptic medication use and had no history of nephrolithiasis or hearing loss. The patient had no other known medical comorbidities and was taking no medications.
Family history was notable for hypothyroidism in his mother and hyperthyroidism in his paternal grandfather. His brother and sister are healthy, and the patient has no children.
On examination, the patient had a BMI of 24.6 kg/m2. He was alert and in no distress. He had prominence of the right brow and left cheek and significant bony deformities of all extremities. No dental abnormalities, cushingoid features or tremors were noted.
The patient's reflexes were normal and symmetric. The thyroid gland was estimated at 25 grams with a prominent right thyroid lobe without discrete nodularity. The patient had a flat hyperpigmented brown circular lesion with irregular borders immediately distal to the right knee.
Laboratory testing results
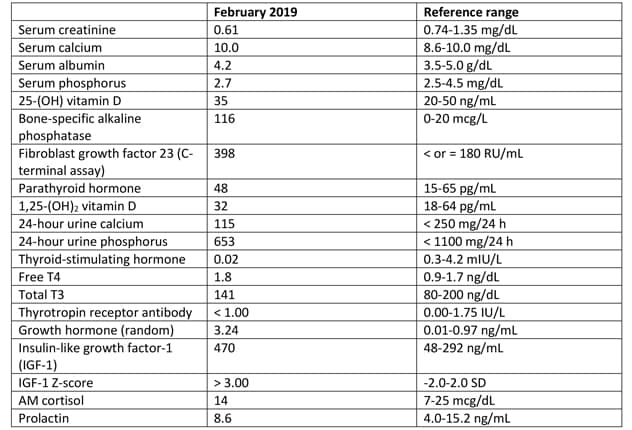
Laboratory testing results
Laboratory studies identified elevated bone-specific alkaline phosphatase and fibroblast growth factor 23.
Polyostotic fibrous dysplasia involving the calvaria and mandible
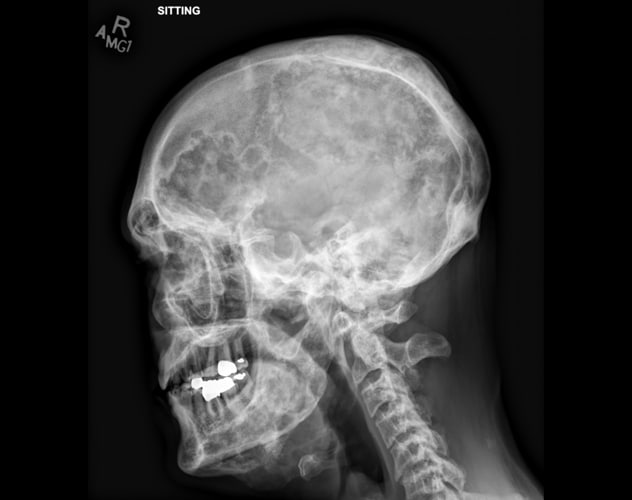
Polyostotic fibrous dysplasia involving the calvaria and mandible
Skeletal survey demonstrates extensive, advanced changes of polyostotic fibrous dysplasia involving multiple bones, including the calvaria and mandible. Typical imaging features include lucent intramedullary lesions with ground-glass opacity and mild bone expansion.
Polyostotic fibrous dysplasia involving the humeri
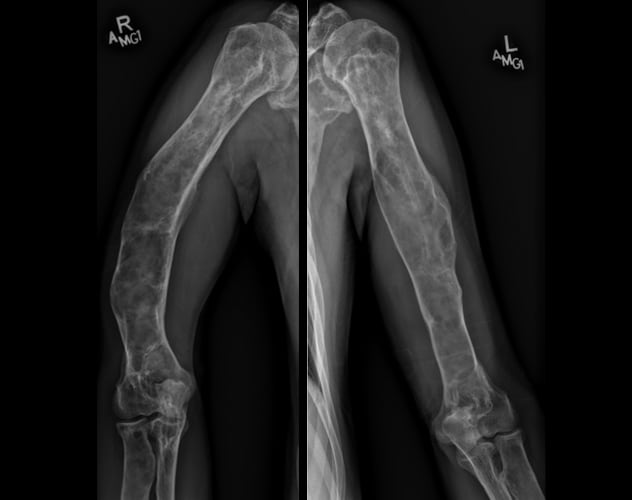
Polyostotic fibrous dysplasia involving the humeri
Skeletal survey demonstrates extensive, advanced changes of polyostotic fibrous dysplasia involving multiple bones, including the humeri.
Polyostotic fibrous dysplasia involving the forearms
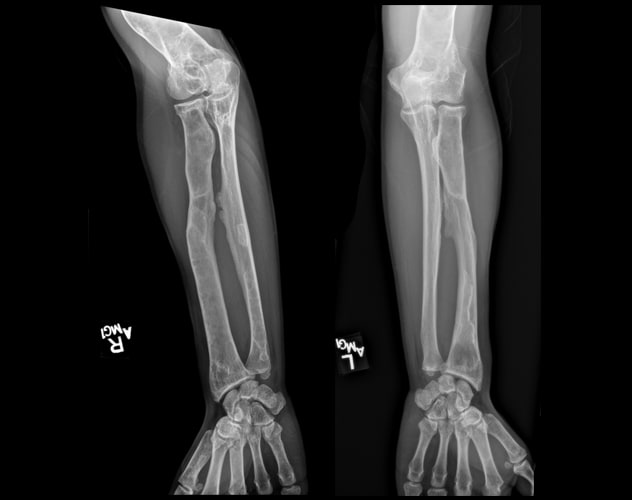
Polyostotic fibrous dysplasia involving the forearms
Skeletal survey demonstrates extensive, advanced changes of polyostotic fibrous dysplasia involving multiple bones, including the forearms.
MRI pituitary without (A) and with (B) intravenous contrast
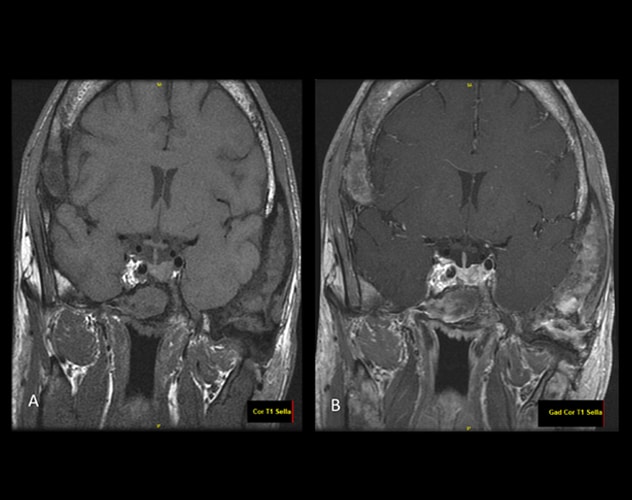
MRI pituitary without (A) and with (B) intravenous contrast
Post-contrast imaging demonstrates a focus with heterogeneous enhancement involving either the left side of the pituitary or the left side of the sella just lateral to the mildly displaced pituitary gland. This may represent a microadenoma, but the findings are not entirely characteristic of an adenoma and the changes may simply be lateral to the pituitary gland itself.
Laboratory studies identified elevated bone-specific alkaline phosphatase and fibroblast growth factor 23 (FGF23) as a result of extensive fibrous dysplasia. In addition, mild hyperthyroidism with negative thyrotropin receptor antibody and elevated insulin-like growth factor-1 (IGF-1) were present. An oral glucose suppression test confirmed growth hormone excess.
Skeletal survey identified diffuse polyostotic fibrous dysplasia. MRI pituitary demonstrated extensive changes of fibrous dysplasia involving the calvaria and skull base and a possible pituitary microadenoma.
Neck ultrasound revealed confluent nodularity in the right lobe with multiple small solid isoechoic nodules. Thyroid uptake and scan were consistent with autonomously functioning thyroid nodules without any cold nodules.
Dual-energy X-ray absorptiometry demonstrated:
- Left distal 1/3 radius Z-score of -1.5
- Left femoral neck Z-score of -1.5
- Left total hip Z-score of -1.3
- L1-L4 Z-score of -1.1
Diagnosis: McCune-Albright syndrome
The patient was diagnosed with McCune-Albright syndrome (MAS), given his polyostotic fibrous dysplasia, cafe au lait macule (skin lesion distal to the right knee), growth hormone excess and autonomously functioning thyroid nodules.
"Remarkably, despite his severe imaging findings, our patient denied significant bone pain; we thus did not pursue treatment with a bisphosphonate. He received definitive hyperthyroidism treatment with 20 mCi of I-131, which proved successful with subsequent development of hypothyroidism requiring levothyroxine replacement," says Peter J. Tebben, M.D., a pediatric endocrinologist at Mayo Clinic in Rochester, Minnesota. "We recommended medical therapy for acromegaly using intramuscular octreotide acetate depot injections, which eventually achieved normal IGF-1 levels."
Discussion
MAS occurs due to post-zygotic gain of function alterations in the GNAS gene, which encodes the α-subunit of the Gs signaling protein. These mutations ultimately lead to constitutive cyclic apical membrane proteins signaling. Patients with GNAS variants have significant phenotypic variability ranging from asymptomatic monostotic fibrous dysplasia (FD) to severe bone disease with multiple endocrinopathies.
In Orphanet Journal of Rare Diseases in 2019, the FD/MAS international consortium defined MAS as the combination of FD (monostotic or polyostotic) and at least one extra-skeletal manifestation or the presence of at least two extra-skeletal features. Extra-skeletal manifestations include:
- Hyperpigmented skin lesions with irregular borders that typically do not cross the midline (cafe au lait macules)
- Gonadal involvement that may manifest as peripheral (gonadotropin-independent) precocious puberty, recurrent ovarian cysts in girls and women or testicular lesions in boys and men
- Thyroid disease with multiple thyroid nodules that can be autonomously functioning
- Growth hormone excess
- Hypercortisolism in the neonatal period
"MAS is a clinical diagnosis and in cases of diagnostic uncertainty genetic testing can be pursued. It is important to note that because MAS is a mosaic disorder, affected tissue is required to identify variants in the GNAS gene," says Jacob D. Kohlenberg, M.D., a resident in Endocrinology, Diabetes, Metabolism, and Nutrition at Mayo Clinic in Rochester, Minnesota. The estimated prevalence of MAS is between 1/100,000 and 1/1,000,000, as reported in research published in Orphanet Journal of Rare Diseases in 2008.
Polyostotic fibrous dysplasia
In MAS, FGF23 associated hypophosphatemia is a surrogate measure of the severity of fibrous dysplasia and renal phosphate wasting is a predictor of future fracture. Bone pain may be present due to FD itself or due to FGF23 mediated hypophosphatemia or both. To treat bone pain, it is essential to address hypophosphatemia if present and optimize the serum 25-hydroxy vitamin D concentration.
First line pharmacologic therapy for bone pain is acetaminophen. For persistent moderate to severe bone pain, an intravenous bisphosphonate such as pamidronate or zoledronate can be used. However, it is unclear if bisphosphonates reduce FD lesion size or progression.
Acromegaly
The prevalence of acromegaly in patients with MAS is 20% to 30%. In patients with MAS, the primary change underlying acromegaly is somatotroph hyperplasia involving the entire pituitary gland, with or without a somatotroph adenoma. Notably, acromegaly is typically associated with skull base FD, which carries an increased risk of hemorrhage due to increased vascularity.
A review of 112 patients with MAS and acromegaly published in The Journal of Endocrinology & Metabolism in 2014 reported that a pituitary adenoma was present in 54% of patients. Postoperative control was achieved in only three of 25 (12%) patients who received surgery: one received radiotherapy preoperatively and the other two received a large or total hypophysectomy.
Because somatotroph hyperplasia involves the entire pituitary gland and surgery can be technically challenging and generally requires performing a total hypophysectomy, medical therapy is preferred as first line treatment. Dopamine agonists may be minimally effective and somatostatin analogues (either as monotherapy or in combination with a dopamine agonist) were effective in lowering GH/IGF-1 in about one-third of patients.
Pegvisomant has been reported to successfully normalize IGF-1 in most patients. Limited data suggests that pituitary radiation is often unsuccessful. Furthermore, malignant transformation of FD has been reported in a small number of cases, which also limits the utility of radiotherapy.
Thyrotoxicosis due to a toxic multinodular goiter
In MAS, patients can have a toxic multinodular goiter and there is an increase in deiodinase activity with an increase in the T3 to T4 ratio.
Learning points
- Patients with fibrous dysplasia, whether monostotic or polyostotic, should be evaluated for possible endocrinopathy and MAS.
- Bone pain can be due to chronic FGF23 mediated hypophosphatemia or the fibrous dysplasia lesions. After correction of hypophosphatemia and vitamin D deficiency (if present) an intravenous bisphosphonate may be helpful for some with moderate to severe bone pain.
- In MAS, the primary etiology underlying acromegaly is somatotroph hyperplasia involving the entire pituitary gland, with or without a somatotroph adenoma. Therefore, medical therapy is preferred as first line treatment.
For more information
Javaid MK, et al. Best practice management guidelines for fibrous dysplasia/McCune-Albright syndrome: A consensus statement from the FD/MAS international consortium. Orphanet Journal of Rare Diseases. 2019;14:139.
Dumitrescu CE, et al. McCune-Albright syndrome. Orphanet Journal of Rare Diseases. 2008;3:12.
Salenave S, et al. Acromegaly and McCune-Albright syndrome. The Journal of Clinical Endocrinology and Metabolism. 2014;99:1955.