Sept. 15, 2018
Lynch syndrome (LS) is a hereditary cancer predisposition syndrome caused by inactivating mutations in DNA mismatch repair (MMR) genes. Germline mutations in the MSH6 MMR gene account for approximately 18 percent of LS cases.
Axial and coronal CT images of right adrenal mass
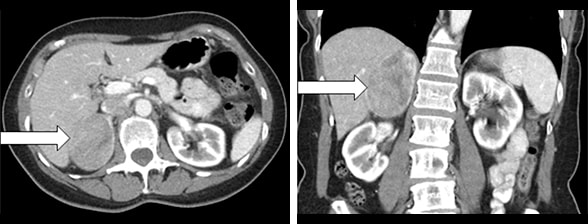
Axial and coronal CT images of right adrenal mass
Figure 1. Axial CT image (left panel) and coronal CT image (right panel) showing a 6.0-by-5.1-by-7.8-cm right adrenal mass (arrows).
A 65-year-old woman with a past medical history of hypertension was diagnosed with LS due to an MSH6 germline mutation as part of family testing. (Her sister, who had a history of colon and endometrial cancers, was the LS proband in the family). The woman was evaluated with a colonoscopy, which was normal. Transvaginal ultrasound was performed to evaluate uterine bleeding; it showed a retroverted uterus with endometrial thickening and uterine fibroids. The ultrasound study detected a heterogeneous echogenic mass within the right upper quadrant of the abdomen. The patient subsequently underwent dilation and curettage of endometrial hyperplasia and pathology was benign. An abdominal CT scan showed a heterogeneous right adrenal mass, 6.0-by-5.1-by-7.8 cm.
Results of biochemical testing
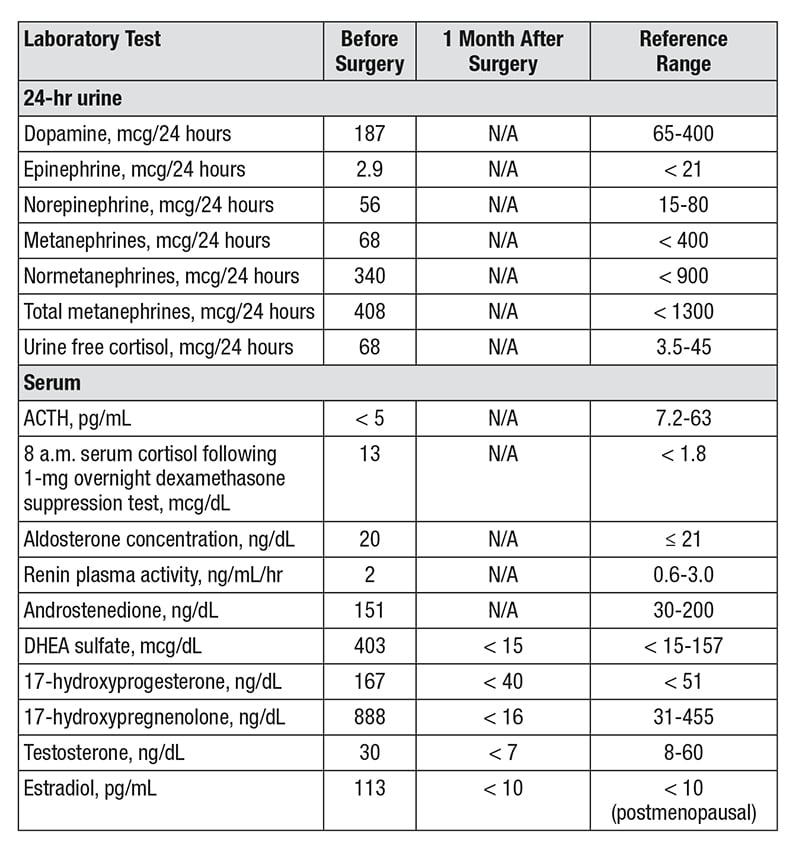
Results of biochemical testing
Results of biochemical testing demonstrate androgen, estrogen and corticotropin-independent cortisol excess.
The woman was referred to Mayo Clinic for further evaluation of the right adrenal mass. The patient had noted symptoms of fatigue, loss of appetite and a 3-pound weight loss over the prior two weeks. There was no history of any signs or symptoms of adrenal hormone excess. On physical examination she did not have cushingoid features, acne or hirsutism. However, biochemical testing demonstrated androgen, estrogen and corticotropin-independent cortisol excess.
Final pathology demonstrates adrenal oncocytic ACC
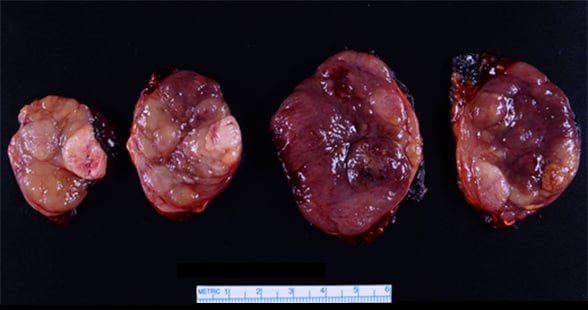
Final pathology demonstrates adrenal oncocytic ACC
Gross pathology serial cut sections of a 9.2-cm right adrenocortical carcinoma (ACC).
Based on the clinical, biochemical and imaging presentation, adrenal cortical carcinoma (ACC) was suspected and an open right adrenalectomy was performed. Final pathology demonstrated a 9.2-by-5.9-by-4.8-cm adrenal oncocytic ACC with focal extracapsular extension into periadrenal adipose tissue, Ki-67 index of 15 percent and a peak mitotic count of 40 mitoses in 50 high-powered fields.
Postoperatively, the patient was treated with glucocorticoid replacement therapy; treatment with mitotane was started six weeks postoperatively. During the subsequent 18 months of follow-up, the patient remains in remission as demonstrated by negative imaging and normalization of steroid precursors.
Discussion
ACC occurs in up to 5 percent of patients with adrenal tumors. ACC occurs most frequently in the fifth to sixth decade of life and demonstrates a female to male predominance of 2.5-to-1. Although most ACCs are sporadic, they can also occur as a part of syndromes such as Li-Fraumeni syndrome, Beckwidth-Wiedemann syndrome, multiple endocrine neoplasia, type 1, familial adenomatous polyposis, neurofibromatosis 1, and LS.
In a study published in the Journal of Clinical Oncology in 2013, the prevalence of LS in patients with ACC was 3.2 percent and higher than in the general population (0.2 percent) and comparable to the prevalence of LS in patients with colorectal cancer (2-4 percent) and endometrial cancer (1-5 percent). The patient described herein had no other manifestations of LS.
ACCs are typically large tumors with a median size greater than 10 cm. The diagnosis of ACC is based on clinical presentation and imaging characteristics of adrenal mass, but remains challenging due to suboptimal accuracy of available tests. As outlined in an article published in Current Opinion in Endocrinology, Diabetes and Obesity in 2017, urine steroid metabolomics is a promising tool to diagnose ACC and is currently under validation. It is important to note that when ACC is suspected, the clinician should avoid biopsy and proceed with adrenalectomy.
Surgical resection is the mainstay of treatment for ACC with a goal to achieve a R0 resection. Further management depends on the stage of ACC and prognostic markers derived from pathology examination.
In the patient under discussion who presented with ACC associated with extracapsular extension and a Ki-67 of 15 percent and underwent a R0 resection, mitotane therapy was recommended and initiated. The patient was monitored at every three-month clinical visit for evaluation of mitotane efficacy and side effects, and with cross-sectional imaging for the first year with a plan to decrease the frequency of follow-up visits in the second year.
For more information
Raymond VM, et al. Adrenocortical carcinoma is a Lynch syndrome-associated cancer. Journal of Clinical Oncology. 2013;31:3012.
Bancos I, et al. Diagnosis of a malignant adrenal mass: The role of urinary steroid metabolite profiling. Current Opinion in Endocrinology, Diabetes and Obesity. 2017;24:200.