Descripción general
Tumor de Wilms
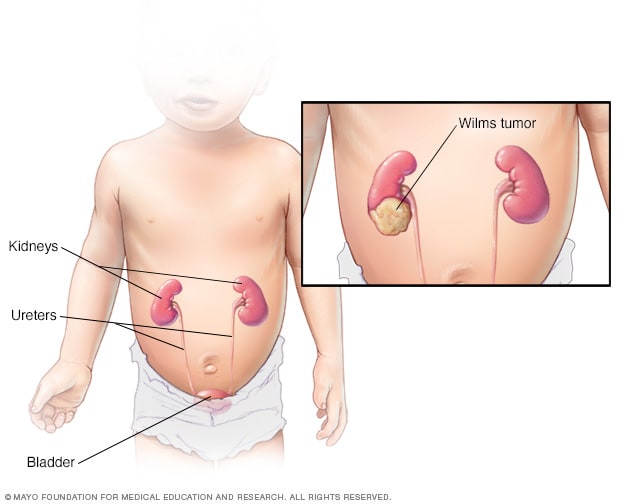
Tumor de Wilms
El tumor de Wilms es el tipo de cáncer de riñón más común en niños. Los riñones forman parte del sistema urinario, el cual se encarga de eliminar desechos del cuerpo a través de la orina. El sistema urinario también comprende los uréteres, la vejiga y la uretra.
El tumor de Wilms es un cáncer de riñón poco común que afecta principalmente a los niños. También conocido como nefroblastoma, es el tipo de cáncer de riñón más habitual en los niños. Por lo general, el tumor de Wilms afecta a niños de 3 a 4 años. Es mucho menos frecuente a partir de los 5 años, pero puede afectar a niños mayores e incluso a adultos.
El tumor de Wilms se produce principalmente en un solo riñón, pero a veces puede estar en ambos riñones al mismo tiempo.
Con los años, los avances en el diagnóstico y tratamiento del tumor de Wilms han mejorado mucho el pronóstico para los niños con esta enfermedad. Con el tratamiento, la perspectiva para la mayoría de los niños que tienen tumor de Wilms es buena.
Productos y servicios
Síntomas
Los síntomas del tumor de Wilms varían en gran medida. Algunos niños con tumor de Wilms parecen no presentar ningún síntoma, otros presentan uno o más de los siguientes:
- Un tumor que se puede palpar en el área estomacal.
- Hinchazón en el área estomacal.
- Dolor en el área estomacal.
Otros síntomas pueden incluir los siguientes:
- Fiebre
- Sangre en la orina.
- Nivel bajo de glóbulos rojos, también conocido como anemia.
- Presión arterial alta.
Cuándo consultar al médico
Programa una cita médica con el proveedor de atención médica de tu hijo si tiene síntomas que te preocupan. El tumor de Wilms es poco frecuente, por lo que es probable que algo más esté causando los síntomas. Sin embargo, es importante revisar cualquier inquietud.
Causas
No está claro cuál es la causa del tumor de Wilms.
El cáncer comienza cuando las células desarrollan cambios en su ADN. El ADN de una célula contiene instrucciones que le indican a la célula lo que debe hacer. Los cambios les indican a las células que crezcan o se multipliquen rápidamente. Las células cancerosas permanecen vivas mientras que las sanas mueren como parte de su ciclo de vida natural. En el caso del tumor de Wilms, los cambios producen células extra en el riñón que forman un tumor.
En raras ocasiones, los cambios en el ADN que se transmiten de padres a hijos pueden aumentar el riesgo de tumor de Wilms.
Factores de riesgo
Algunos de los factores que pueden aumentar el riesgo de padecer tumor de Wilms son los siguientes:
- Ser de piel negra. En Norteamérica y Europa, los niños de piel negra tienen un riesgo levemente mayor de presentar un tumor de Wilms que los niños de otras razas. Al parecer, los niños de origen asiático-americano tienen un riesgo menor que los niños de otras razas.
- Tener antecedentes familiares de tumor de Wilms. El riesgo de contraer la enfermedad es mayor si alguien de la familia tuvo un tumor de Wilms.
El tumor de Wilms ocurre más comúnmente en los niños que presentan ciertas afecciones al nacer, como las siguientes:
- Aniridia. En este trastorno, el iris, que es la parte coloreada del ojo, se forma de manera parcial o está ausente.
- Hemihipertrofia. La hemihipertrofia es una afección por la que una parte o un lado del cuerpo es más grande que el otro.
El tumor de Wilms puede producirse como parte de síndromes poco frecuentes, entre ellos, los siguientes:
- Síndrome WAGR. Este síndrome se presenta con tumor de Wilms, aniridia, problemas en los genitales y el sistema urinario, y discapacidades intelectuales.
- Síndrome de Denys-Drash. Este síndrome se presenta con tumor de Wilms, enfermedad renal y pseudohermafroditismo masculino. Un niño con pseudohermafroditismo masculino tiene genitales que no son claramente masculinos.
- Síndrome de Beckwith-Wiedemann. Los niños que tienen este síndrome suelen ser mucho más grandes que lo habitual, lo que se conoce como macrosomia. A causa de este síndrome, puede ocurrir que los órganos de la zona abdominal sobresalgan de la base del cordón umbilical y que la lengua y los órganos internos sean grandes y las orejas tengan una forma inusual.
Prevención
El tumor de Wilms no puede prevenirse.
Si un niño tiene algunas de las afecciones que aumentan el riesgo de desarrollar el tumor de Wilms, un proveedor de atención médica puede sugerir que se realicen ecografías de riñón periódicas para detectar cualquier anomalía en los riñones. Si bien estos exámenes de detección no pueden prevenir el tumor de Wilms, pueden ayudar a detectar la enfermedad en una etapa temprana.