Oct. 22, 2019
A 49-year-old white female presented to the bone clinic at Mayo Clinic in Rochester, Minnesota, with a history of intermittent joint pains affecting her hands, feet and elbows since her 20s, periarticular growths, and bilateral conductive hearing loss. There was no history of fractures, renal dysfunction, nephrolithiasis or hypercalcemia.
Laboratory data for case patient (proband) and her sister
Examination revealed normal midparental height, nondysmorphic body habitus, Heberden's and Bouchard's nodes in the fingers, and hammertoe deformities of the feet. Laboratory workup was significant for a low serum phosphorus; elevated parathyroid hormone (PTH); elevated 1,25(OH)2D; and reduced renal tubular reabsorption of phosphorus, or TRP. Dual energy X-ray absorptiometry showed osteopenia (worst femur neck T-score: -2.3), and parathyroid sestamibi scan was nonlocalizing.
The patient was diagnosed with normocalcemic primary hyperparathyroidism (PHPT) and her symptoms were treated with vitamin D and calcium supplementation. However, a year later, she underwent removal of left and right superior parathyroid glands at another institution.
The patient returned to Mayo Clinic at age 52 with intermittent symptomatic hypocalcemia, persistent hypophosphatemia and a normal PTH. Her family history was notable for a sister who was deaf and had severe hypophosphatemic rickets, periarticular mineral deposition, and a history of normocalcemic primary hyperparathyroidism with multigland parathyroidectomy.
C-terminal fibroblast growth factor 23 (FGF23) was in the normal range, but with the concern for hereditary non-PTH mediated hypophosphatemia, intact FGF23 was tested and found to be elevated.
Plain radiograph demonstrating bowing of right femur
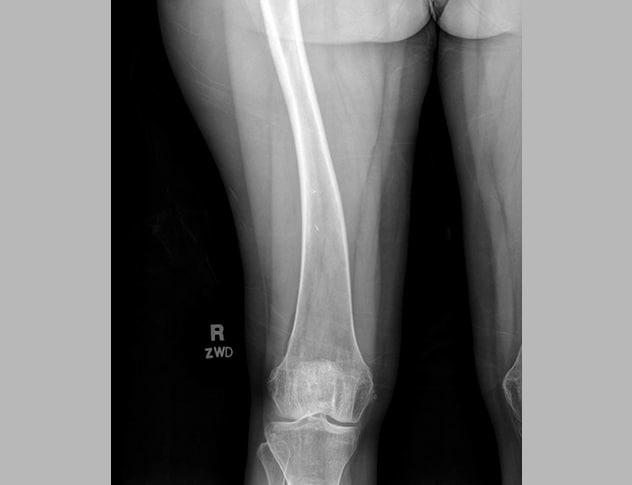
Plain radiograph demonstrating bowing of right femur
Detailed bone radiograph demonstrates bowing of right femur.
Plain radiographs demonstrating periarticular calcifications
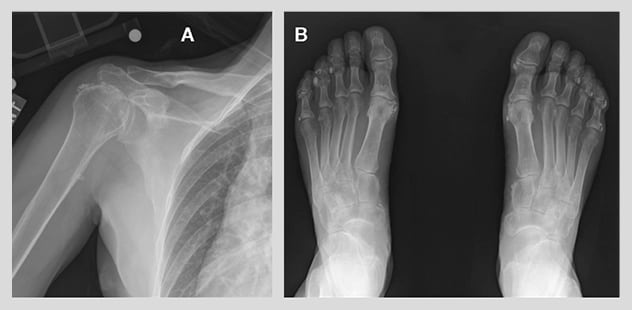
Plain radiographs demonstrating periarticular calcifications
Detailed bone radiographs demonstrate periarticular calcifications in the right shoulder (A) and both feet (B).
Family tree ENPP1 mutation family pedigree
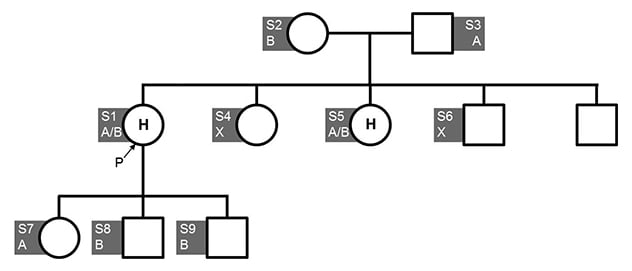
Family tree ENPP1 mutation family pedigree
Family tree ENPP1 mutation family pedigree. H = Hypophosphatemic rickets, P = Proband, S = Subject number, ENPP1 mutation: HET c.323G>T p.Cys108Phe (A) and HET c.1441C>T p.Arg481TRP (B),
X (none).
Detailed bone radiographs demonstrated bowing of bilateral femurs and periarticular calcifications in hands, elbows, knees, shoulders and feet.
The patient was evaluated by Medical Genetics staff and underwent whole-exome sequencing that identified two variants in the ENPP1 gene:
- Heterozygous (HET) c.323G>T p.Cys108Phe (inherited from father)
- HET c.1441C>T p.Arg481TRP (inherited from mother)
No PHEX, FGF23 or DMP1 mutations were identified, hence ruling out other forms of hereditary hypophosphatemia. The sister who had a similar but more severe phenotype had the same biallelic mutations and each of the patient's three children had monoallelic mutations.
Vascular health screening demonstrated increased carotid intima-media thickness but no vascular calcification. The patient was treated with calcium, vitamin D-3 and calcitriol, which led to improvement in serum calcium and phosphorus.
Discussion
Hereditary hypophosphatemia is a form of FGF23-mediated hypophosphatemia categorized as X-linked hypophosphatemia, autosomal dominant hypophosphatemic rickets or the much rarer autosomal recessive hypophosphatemic rickets (ARHR) types 1 and 2. ARHR2 is associated with deficiency of the ENPP1 enzyme, which generates pyrophosphate (PPi) from adenosine triphosphate, but its association with FGF23 is unclear. The clinical features of ARHR2 in adults include:
- Periarticular calcifications with a waxing and waning clinical course over years
- History of rickets
- Conductive hearing loss
Rickets and hypophosphatemia are mediated by FGF23 produced by bones, which decreases renal phosphate reabsorption and decreases 1-alpha hydroxylase activity. Hence a patient with hypophosphatemia, low or normal PTH, and low or inappropriately normal 1,25(OH)2D for the level of hypophosphatemia should raise concern for FGF23-mediated hypophosphatemia.
As reported in Human Mutation in 2016, periarticular calcifications in ARHR2 are likely due to PPi deficiency, because PPi inhibits hydroxyapatite crystal deposition as well as arterial intima-media proliferation. Periarticular enthesopathy has not been previously described in patients with ARHR2 but is seen in ENPP1 knockout rodent models, as reported in Bone in 2012; we identified that the proband's sister had enthesopathy.
PHPT has not been previously noted in patients with ENPP1 variants. Reduced 1,25(OH)2D mediated through increased FGF23 downregulation of 1-alpha hydroxylase activity over time can lead to chronic parathyroid stimulation, which could contribute to PHPT. Our patients confirm the observation that C-terminal FGF23 can be normal but may be inappropriately so for the extent of hypophosphatemia; however, intact FGF23 is usually elevated with biallelic ENPP1 genetic variants.
Given the challenges of diagnosing hereditary hypophosphatemia based on clinical and biochemical findings, gene sequencing should be considered. Inactivating ENPP1 variants are associated with generalized arterial calcification of infancy (GACI) and less often ARHR2.
Variable disease severity is reported, with infants presenting with the severe GACI while children and adults present with ARHR2, as reported in American Journal of Human Genetics in 2010 and Nature Genetics in 2003. However, such pathogenic variants are extremely rare, with only six adults with ARHR2 caused by inactivating ENPP1 variants, also reported in Nature Genetics in 2003.
Hence, our patient and her sister with inactivating compound heterozygous ENPP1 variants, one of which has been associated with GACI and reported in Nature Genetics in 2003, in addition to a novel variant not previously identified, add to the limited knowledge. This kinship suggests that compound heterozygous carriers of the HET c.1441C>T p.Arg481TRP pathogenic variant develop less severe disease than homozygous carriers, and that the protein generated by the allele altered by the HET c.323G>T p.Cys108Phe variant retains some functionality of ENPP1 enzyme.
ENPP1 may perform other functions in addition to PPi generation that may contribute to the uncommon phenotype observed. It is significant that both compound heterozygous subjects developed PHPT, a condition affecting a tissue where ENPP1 is also expressed. Homozygous biallelic mutations known to cause GACI have been associated with death in early childhood, whereas compound heterozygous mutations demonstrate survival beyond the critical early childhood period in some families.
Identification of ENPP1 mutations causing GACI or ARHR2 has increased clinical relevance due to the recent development of ENPP1 enzyme replacement therapy (INZ-701), which has demonstrated restoration of PPi to normal levels reducing complications associated with ENPP1 deficiency in rodents, as reported in Nature Communications in 2015.
Key points
- In hereditary hypophosphatemia, FGF23 levels should be interpreted relative to the degree of hypophosphatemia, keeping in mind the limitations of the clinically available C-terminal assay.
- Loss of function variants in genes encoding the enzyme ENPP1 cause GACI and ARHR2 as part of a phenotypic spectrum.
- Monoallelic ENPP1 variants may cause subtle biochemical and clinical findings.
- Whole-exome sequencing can be useful in the diagnosis and detection of new causative mutations of rare bone and mineral disorders.
For more information
Stella J, et al. Effects of different variants in the ENPP1 gene on the functional properties of ectonucleotide pyrophosphatase/phosphodiesterase family member 1. Human Mutation. 2016;37:1190.
Mackenzie NCW, et al. New insights into NPP1 function: Lessons from clinical and animal studies. Bone. 2012;51:961.
Lorenz-Depiereux B, et al. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. American Journal of Human Genetics. 2010;86:267.
Rutsch F, et al. Mutations in ENPP1 are associated with 'idiopathic' infantile arterial calcification. Nature Genetics. 2003;34:379.
Albright RA, et al. ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nature Communications. 2015;6:10006.