June 09, 2018
A 28-year-old man was referred for further evaluation of a right adrenal mass and a right pleural mass. He was in overall good health until 1.5 years ago, when he was noted to have a blood pressure reading of greater than 170/90 mm Hg on more than two occasions at his occupational health facility. He was started on antihypertensive medications.
Two months later, screening laboratory studies showed an elevated hemoglobin A1C (7.5 percent) and the patient was diagnosed with type 2 diabetes mellitus and treated with metformin. Medications were titrated to achieve optimal blood pressure, blood glucose and lipid control. He was on six new medications, including two antihypertensives.
As part of his occupational screen, a chest X-ray was performed one year ago, which showed a right pleural mass measuring 4 cm by 3 cm. A three-month follow-up chest X-ray showed growth of the mass to 5.3 cm by 3.1 cm.
Due to the concern for malignancy, a fluorodeoxyglucose positron emission tomography (FDG-PET) scan was performed and this detected minimal fluorodeoxyglucose uptake in a right adrenal mass measuring 7.3 cm in largest diameter, a hypermetabolic pleural mass, a presacral mass and multiple soft tissue masses that were not hypermetabolic. Subsequent biopsy of the pleural mass confirmed this to be a peripheral nerve sheath tumor.
On referral to Mayo Clinic, he was asymptomatic. He denied paroxysms, palpitations, presyncope or syncopal events. He did not smoke, use illicit drugs or drink alcohol. Physical examination was significant for diffuse freckling over his shoulders, chest, axillary and inguinal areas. He had four cafe au lait spots on his trunk and several cutaneous and subcutaneous soft tissue lesions over his forehead, neck and extremities. His father had a history of hypertension diagnosed in his 50s.
Pre- and postoperative test results
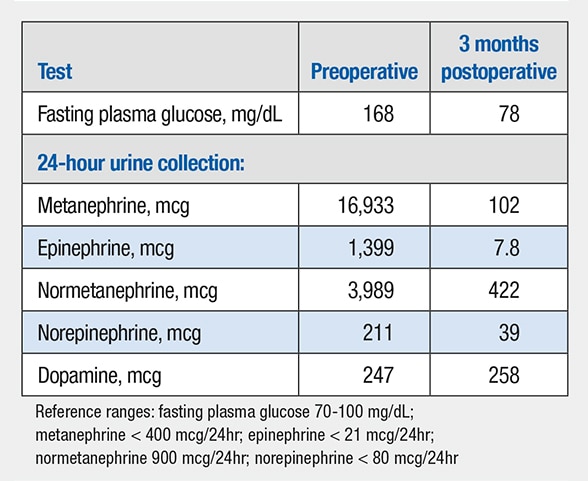
Pre- and postoperative test results
Preoperative and three months postoperative laboratory tests.
Right adrenal pheochromocytoma and pleural-based nerve sheath tumor
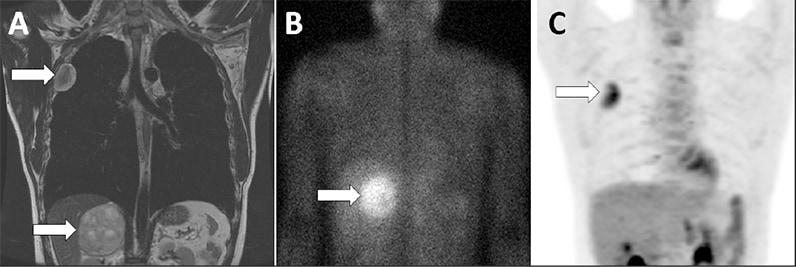
Right adrenal pheochromocytoma and pleural-based nerve sheath tumor
Coronal images of the chest and abdomen with arrows highlighting the right adrenal pheochromocytoma and the pleural-based nerve sheath tumor on MRI (A), 123-I-MIBG scan (B) and FDG-PET scan (C).
His presentation and physical examination findings were consistent with neurofibromatosis 1 (NF1). The levels of fractionated metanephrines and catecholamines in a 24-hour urine collection were elevated. Given the multiple masses found on the FDG-PET scan, additional imaging was obtained and included I-123 metaiodobenzylguanidine (MIBG) scintigraphy and MRI of the chest and abdomen.
MIBG uptake was limited to the right adrenal mass. The FDG-PET scan showed activity in the pleural mass but not the adrenal mass.
Preoperative adrenergic blockade included phenoxybenzamine, metoprolol and metyrosine. The patient underwent an open right adrenalectomy without complication. Final pathology showed a 9.8-cm pheochromocytoma encompassing the entire right adrenal gland with negative margins. Genetic analysis was positive for heterozygous splicing mutation in the NF1 gene.
Pheochromocytoma is a rare neoplasm with an estimated annual incidence of 0.8 per 100,000 person-years. It is the cause of hypertension in less than 0.2 percent of patients. Genetic causes of pheochromocytoma account for 30 percent of all cases. Hereditary pheochromocytomas tend to occur at a younger age (for example, younger than 45 years) and are usually inherited in an autosomal dominant fashion.
The three most common hereditary causes of adrenal pheochromocytoma are von Hippel-Lindau disease, multiple endocrine neoplasia, type 2, and NF1. Approximately 3 percent of patients with NF1 will have a catecholamine-secreting tumor, most commonly a unilateral benign adrenal pheochromocytoma or periadrenal paraganglioma.
A Mayo Clinic study published in Clinical Endocrinology in 2017 found that only 58 percent of catecholamine-secreting tumors in patients with NF1 were symptomatic, and of those who were symptomatic, hypertension was the most common symptom (76 percent).
Bilateral adrenal pheochromocytoma was found in seven (17 percent) of the 41 patients with NF1; 7.3 percent of patients had recurrent or metastatic disease. In the seven patients with bilateral adrenal pheochromocytoma, four underwent cortical-sparing surgery, negating the need for glucocorticoid and mineralocorticoid replacement.
Asymptomatic pheochromocytoma is quite common in the setting of NF1. It has been suggested that the prevalence of pheochromocytoma in NF1 may be as high as 6.6 percent when case detection testing is routinely performed. It is recommended that biochemical case detection testing for pheochromocytoma be performed every three years in patients with NF1.
Key message
NF1 has a high prevalence of asymptomatic pheochromocytoma with hypertension being the most common symptom. Biochemical case detection should be performed every three years in asymptomatic patients with NF1.
For more information
Gruber LM, et al. Pheochromocytoma and paraganglioma in patients with neurofibromatosis type 1. Clinical Endocrinology. 2017;86:141.