Diagnóstico
Biopsia de hígado
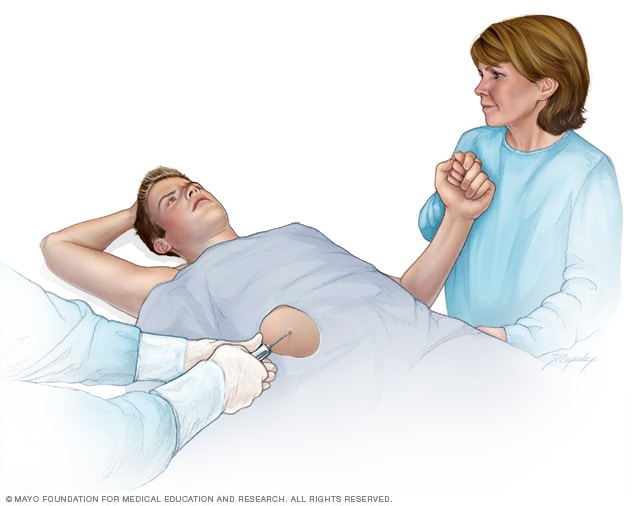
Biopsia de hígado
Una biopsia de hígado extrae una pequeña muestra de tejido hepático para realizar pruebas de laboratorio. La biopsia de hígado se realiza con mayor frecuencia introduciendo una aguja fina a través de la piel hasta el hígado.
El diagnóstico de la enfermedad de Wilson puede ser difícil porque sus síntomas son similares a los de otras enfermedades hepáticas, como la hepatitis. Además, los síntomas pueden aparecer lentamente con el tiempo. Los cambios en el comportamiento que aparecen progresivamente pueden ser, en particular, difíciles de vincular con la enfermedad de Wilson.
Criterios de diagnóstico
Los profesionales de atención médica diagnostican la enfermedad de Wilson en función de una combinación de síntomas, valores de laboratorio y obtención de imágenes o pruebas genéticas. Una herramienta de puntuación estructurada, como los criterios diagnósticos de Leipzig, puede ayudar a los profesionales de atención médica a combinar estos síntomas y resultados de las pruebas para confirmar o descartar la enfermedad de Wilson. La herramienta asigna puntos en función de los síntomas y los resultados de las pruebas. Una puntuación de Leipzig de 4 o más, por ejemplo, significa que es muy probable que se trate de la enfermedad de Wilson.
Las pruebas y los procedimientos pueden consistir en lo siguiente:
Análisis de sangre y de orina
Los análisis de sangre pueden indicar si tu hígado funciona correctamente y medir los niveles de ceruloplasmina, una proteína que se adhiere al cobre en la sangre. Los análisis de sangre también pueden verificar el nivel de cobre en la sangre. Es posible que tu profesional de atención médica quiera medir la cantidad de cobre eliminado en la orina durante un período de 24 horas.
- Ceruloplasmina. Esta prueba mide los niveles de una proteína que se une al cobre en la sangre. En la enfermedad de Wilson, los niveles de ceruloplasmina suelen ser bajos. Lo que se considera bajo depende del laboratorio específico, pero, por lo general, se considera bajo un nivel inferior a 20 miligramos por decilitro (mg/dl) o 0,2 gramos por litro (g/l). Los niveles son bajos debido a que la mutación genética del ATP7B que causa la enfermedad de Wilson impide que el cobre se una correctamente a la proteína ceruloplasmina. Algunas personas con la enfermedad de Wilson no tienen niveles bajos de ceruloplasmina. Puedes tener niveles bajos de ceruloplasmina sin tener la enfermedad de Wilson. Por ejemplo, una enfermedad hepática causada por otro motivo puede reducir los niveles de ceruloplasmina.
- Cobre sérico. Esta prueba determina el nivel de cobre en la sangre. La prueba por sí sola no se usa para diagnosticar la enfermedad de Wilson. En la enfermedad de Wilson, los niveles totales de cobre sérico suelen ser bajos debido a los bajos niveles de ceruloplasmina. Sin embargo, el nivel de cobre libre, que es el cobre que no está unido a la ceruloplasmina, suele ser más alto de lo habitual.
- Cobre en la orina durante 24 horas. Es posible que tu profesional de atención médica quiera medir la cantidad de cobre eliminado en la orina durante un período de 24 horas. En la enfermedad de Wilson, este nivel suele ser alto. Lo que se considera alto depende del laboratorio específico, pero, por lo general, se considera alto un nivel superior a valores de 40 a 100 microgramos (mcg) al día.
- Otras pruebas hepáticas. Se pueden hacer otras pruebas para comprobar cómo funciona el hígado. Estas pruebas se denominan análisis de la función hepática. Sin embargo, la enfermedad de Wilson puede causar daño hepático, aunque la función hepática sea normal y también lo sean los resultados de las pruebas. Esto se llama enfermedad hepática compensada.
Examen ocular
Durante un examen ocular, un oftalmólogo puede usar una luz especial llamada lámpara de hendidura para detectar los anillos de Kayser-Fleischer y las cataratas en girasol.
Los anillos de Kayser-Fleischer son de color marrón dorado o cobrizo y aparecen alrededor del borde exterior del iris, la parte con color del ojo. Los anillos los causa la acumulación de cobre en una parte de la membrana de Descemet, una parte de la córnea. La córnea es la capa transparente con forma de cúpula que cubre la pupila y el iris. Los anillos de Kayser-Fleischer se observan en la mayoría de las personas con síntomas neurológicos y en alrededor de la mitad de las personas con síntomas de enfermedad hepática únicamente.
La enfermedad de Wilson también puede causar un tipo especial de catarata, denominada catarata en girasol. Esta catarata está causada por la acumulación de cobre en el lente del ojo.
Biopsia de hígado
En una biopsia de hígado, el profesional de atención médica inserta una aguja delgada a través de la piel hasta llegar al hígado para extraer una pequeña muestra de tejido. A continuación, en el laboratorio, se analiza el tejido para detectar un exceso de cobre. Un nivel de cobre superior a 250 microgramos por gramo (mcg/g) de peso seco del hígado es un signo de la enfermedad de Wilson.
Pruebas genéticas
Un análisis de sangre puede detectar mutaciones en el gen ATP7B, que es el causante de la enfermedad de Wilson. Se conocen cientos de mutaciones en el gen ATP7B. Una de las más comunes es la mutación p.H1069Q. Si tienes el gen mutado que causa la enfermedad de Wilson, tus hermanos e hijos también deben someterse a pruebas genéticas. Detectar la afección a tiempo significa que pueden comenzar el tratamiento antes de que aparezcan los síntomas.
Resonancia magnética
La resonancia magnética es una técnica de obtención de imágenes médicas que utiliza un campo magnético y ondas de radio generadas por computadora para crear imágenes detalladas de los órganos y de los tejidos del cuerpo. En las personas con síntomas neurológicos de la enfermedad de Wilson, una resonancia magnética cerebral puede mostrar cambios en las partes del cerebro afectadas por la acumulación de cobre. El rostro del panda gigante se observa en una resonancia magnética cerebral en alrededor del 10 % al 15 % de las personas con enfermedad de Wilson. Indica que la enfermedad de Wilson ha dañado el mesencéfalo, la parte media del cerebro. El nombre del signo refleja un patrón de colores más claros y más oscuros que se asemejan al rostro de un panda en la imagen de la resonancia magnética.
Afecciones que pueden tener síntomas similares
Cuando se diagnostica la enfermedad de Wilson, los profesionales de atención médica tienen en cuenta otras posibles afecciones que podrían causar síntomas similares. Esto ayuda a garantizar un diagnóstico y un tratamiento correctos.
Todas estas otras afecciones están causadas por diferentes cambios genéticos:
- Hemocromatosis. Al igual que la enfermedad de Wilson, la hemocromatosis puede afectar al hígado. Pero una diferencia entre la enfermedad de Wilson y la hemocromatosis es que la enfermedad de Wilson consiste en una acumulación de cobre. La hemocromatosis consiste en un exceso de hierro.
- Enfermedad de Huntington. Tanto la enfermedad de Wilson como la enfermedad de Huntington pueden causar síntomas similares en el movimiento y el comportamiento. Pero la enfermedad de Wilson se diferencia de la enfermedad de Huntington en que afecta tanto al hígado como al cerebro debido a la acumulación de cobre. La enfermedad de Huntington afecta al cerebro, ya que causa que las células nerviosas en el cerebro se deterioren con el tiempo.
- Aceruloplasminemia. La aceruloplasminemia y la enfermedad de Wilson no están relacionadas, aunque ambas afectan a la proteína ceruloplasmina que se une al cobre en la sangre. La aceruloplasminemia lleva a una acumulación de hierro en el hígado, no de cobre.
- Enfermedad de Menkes. La enfermedad de Menkes también afecta la forma en que el cuerpo usa el cobre. Pero la enfermedad de Wilson no es lo mismo que la enfermedad de Menkes. La enfermedad de Menkes deriva en un nivel bajo de cobre en el hígado y el cerebro. La enfermedad de Wilson deriva en un nivel alto de cobre en el hígado y el cerebro.
- Otras afecciones del metabolismo del cobre o el hierro. Hay otras afecciones hereditarias poco frecuentes que pueden afectar el equilibrio del cobre y el hierro. Sin embargo, tienen causas genéticas diferentes.
Todas estas afecciones las causan cambios genéticos distintos del cambio genético que causa la enfermedad de Wilson. Además, todas tienen efectos diferentes en el cuerpo.
Más información
Tratamiento
En primer lugar, con el fin de tratar la enfermedad de Wilson, un profesional de atención médica puede recomendar medicamentos llamados quelantes del cobre. Estos medicamentos se adhieren al cobre y hacen que los órganos lo liberen en el torrente sanguíneo. Luego, los riñones filtran el cobre y lo eliminan a través de la orina.
El tratamiento posterior consiste en evitar que el cobre vuelva a acumularse. Esto se suele hacer con un medicamento a base de zinc. En el caso de un daño hepático grave, puede ser necesario realizar un trasplante de hígado.
¿Se puede curar la enfermedad de Wilson?
El tratamiento protege al organismo de daños mayores debidos a la enfermedad de Wilson y puede mejorar algunos síntomas. Los medicamentos no curan la enfermedad de Wilson. Un trasplante de hígado no puede curar el cambio genético que causa la enfermedad de Wilson. Sin embargo, un hígado donado no tendría el gen mutado, por lo que procesaría el cobre correctamente sin necesidad de más tratamiento. Si la enfermedad de Wilson se detecta a tiempo y se trata, las personas que la padecen pueden llevar una vida plena y tener una expectativa de vida normal.
Medicamentos
Si tomas medicamentos para la enfermedad de Wilson, deberás seguir tomándolos de por vida, a menos que te realicen un trasplante de hígado.
Quelantes para la eliminación del cobre
Estos medicamentos suelen usarse, en primer lugar, como tratamiento inicial para reducir rápidamente los niveles de cobre:
- Penicilamina (Cuprimine, Depen). La penicilamina, a veces denominada d-penicilamina, es un medicamento que elimina el exceso de cobre del organismo. Puede causar efectos secundarios graves, como problemas cutáneos y renales. También puede ocasionar o empeorar síntomas neurológicos, sobre todo en personas que ya los padecen. Además, puede reducir la capacidad de la médula ósea para producir suficientes glóbulos rojos y plaquetas. Usa la penicilamina con precaución si tienes alergia a este fármaco. Debido a que la penicilamina reduce el efecto de la piridoxina, también conocida como vitamina B6, además tendrás que tomar pequeñas dosis de un suplemento de vitamina B6.
- Trientina (Cuvrior, Syprine). Este es otro medicamento que elimina el exceso de cobre del organismo. Funciona de manera muy similar a la penicilamina, pero suele causar menos efectos secundarios. Sin embargo, los síntomas neurológicos pueden empeorar si tomas trientina.
Terapia con zinc para prevenir la absorción de cobre
El acetato de zinc (Galzin) evita que el cuerpo absorba el cobre de los alimentos que comes. Este medicamento suele usarse como tratamiento de mantenimiento para impedir que el cobre vuelva a acumularse luego de haber recibido un tratamiento inicial con penicilamina o trientina. El acetato de zinc puede usarse como tratamiento principal si no puedes tomar penicilamina ni trientina o si no presentas síntomas. Los efectos secundarios más comunes del acetato de zinc son el malestar estomacal y la diarrea.
Tu profesional de atención médica también puede recomendarte diversas manera de tratar otros síntomas de la enfermedad de Wilson.
Cirugía
Trasplante de hígado con donante vivo
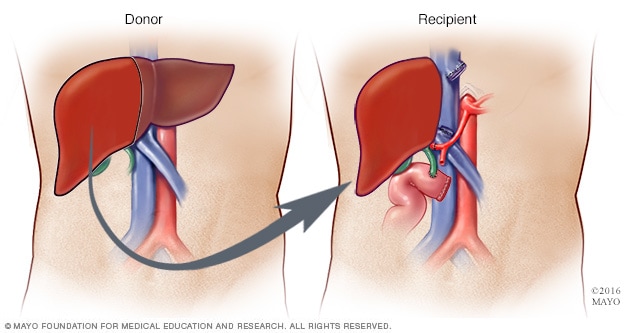
Trasplante de hígado con donante vivo
Durante un trasplante de hígado del donante vivo, los cirujanos extraen entre un 40 % y un 70 % del hígado del donante y lo colocan en el receptor.
Si tu hígado está tan dañado que sufres insuficiencia hepática o si el daño no se puede revertir con medicamentos, es posible que necesites un trasplante de hígado. Durante un trasplante de hígado, el cirujano extirpa el hígado dañado y lo reemplaza por el hígado sano de un donante.
La mayoría de los hígados trasplantados proviene de donantes fallecidos. En algunos casos, el hígado puede provenir de un donante vivo, por ejemplo, de un familiar. En ese caso, el cirujano extirpa el hígado y lo reemplaza con una parte del hígado del donante.
Estilo de vida y remedios caseros
La enfermedad de Wilson se controla con medicamentos de por vida para eliminar y bloquear el exceso de cobre en el organismo, junto con el autocuidado para asegurarte de no consumir demasiado cobre en tu alimentación.
Si te indican que limites el cobre en tu alimentación, esto puede incluir el cobre que se encuentra en el agua, los suplementos y los alimentos.
Agua
Si tu casa tiene tuberías de cobre, deja correr el agua hasta que se enfríe antes de usarla para cocinar o beber. Si utilizas agua de pozo, haz que la analicen para detectar la presencia de cobre o usa un filtro que elimine los oligoelementos.
Suplementos
Asegúrate de evitar los multivitamínicos o suplementos que contienen cobre.
Alimentos
Si padeces la enfermedad de Wilson, habla con tu equipo de atención médica sobre cómo adaptar tu alimentación para cumplir con las restricciones y pautas dietéticas necesarias.
Es probable que tu equipo de atención médica te recomiende limitar el consumo de cobre. Sin embargo, la cantidad adecuada puede depender de la etapa del tratamiento en la que te encuentres, así como de otros factores de salud personales. Por ejemplo, es posible que al inicio te pidan que no consumas más de 1 miligramo (mg) de cobre al día. Más adelante, en un plan de mantenimiento, es posible que te indiquen que puedes consumir una cantidad moderada de cobre, como 2 mg o menos al día.
Alimentos con alto contenido de cobre
Si padeces la enfermedad de Wilson, es posible que te pidan que evites o limites el consumo de los siguientes alimentos:
- Hígado y otras vísceras (entrañas).
- Mariscos y gambas (camarones).
- Carne de cordero.
- Ganso, pato y codorniz.
- Tofu, proteína de soya y leche de soya.
- Frijoles (porotos) secos.
- Setas comestibles.
- Batata (camote) y otras papas con piel.
- Frutas deshidratadas.
- Harina de soya, cereales de salvado y granos de maíz (elote).
- Frutos secos y semillas.
- Chocolate.
- Agua mineral.
Alimentos con bajo contenido de cobre
Tu equipo de atención médica puede indicarte que consumas más de estos alimentos como base de tu dieta si padeces la enfermedad de Wilson:
- Carne blanca de pollo y pavo.
- Bacalao, vieiras, fletán, atún y trucha.
- Huevos, mantequilla y margarina.
- Queso, queso cottage y crema agria.
- Leche de almendras, leche de vaca y yogur, siempre y cuando no sean saborizados.
- Coles de Bruselas, brócoli y repollo.
- Coliflor, espinacas y pimientos verdes.
- Cebollas.
- Hongos shiitake y enoki.
- Manzanas, arándanos azules, cerezas, ciruelas, melones, fresas y sandía.
- Café y bebidas carbonatadas.
Algunos alimentos saludables tienen un nivel moderado de cobre y se pueden consumir en pequeñas cantidades. Pregunta a tu equipo de atención médica qué pautas dietéticas son adecuadas para ti.
Es común tener preguntas o inquietudes sobre si puedes llevar una vida normal con la enfermedad de Wilson. Comenzar a tomar medicamentos de por vida y aprender nuevos hábitos alimenticios puede parecer algo difícil de manejar. Date tiempo para adaptarte y comparte tus inquietudes con el equipo de atención médica. También puede ser útil saber que, con un diagnóstico temprano, un tratamiento médico adecuado y el autocuidado, la mayoría de las personas con la enfermedad de Wilson llevan una vida saludable.
Preparación para la consulta
Es probable que primero consultes a un profesional de atención primaria. Es posible que te remitan a un hepatólogo, que es un profesional de atención médica especializado en el hígado.
Qué puedes hacer
Cuando solicites la cita médica, pregunta si debes hacer algo para prepararte, como modificar tu alimentación antes de los análisis de sangre.
Puede ser útil hacer una lista con lo siguiente:
- Los síntomas y cuándo comenzaron.
- Información personal importante, como situaciones estresantes, otras afecciones y cualquier antecedente familiar de la enfermedad de Wilson.
- Todos los medicamentos, las vitaminas u otros suplementos que tomas y también las dosis.
- Preguntas que quieres hacer a tu profesional de atención médica.
Si es posible, lleva a alguien contigo para que te ayude a recordar la información que recibas.
Si crees que puedes padecer la enfermedad de Wilson o te han dicho que la padeces, es posible que quieras hacerle las siguientes preguntas a tu profesional de atención médica:
- ¿Qué pruebas deben hacerme para confirmar la enfermedad de Wilson?
- ¿Qué datos sobre los antecedentes familiares se necesitan?
- ¿Cómo se trata y se controla la enfermedad de Wilson a largo plazo?
- ¿Qué tratamiento recomienda?
- ¿Cuáles son los efectos secundarios de este tratamiento?
- ¿Cuál es la mejor manera de controlar la enfermedad de Wilson junto con mis otras afecciones?
- ¿Necesito limitar los tipos de alimentos que como?
- ¿Debo acudir a un especialista y, si la respuesta es sí, a cuál?
- ¿Mis hermanos o hijos deben hacerse pruebas para detectar la enfermedad de Wilson?
- ¿A qué tipo de especialista deben acudir los miembros de mi familia?
- ¿Hay algún folleto u otro material impreso que pueda llevarme? ¿Qué sitios web me recomiendan?
No dudes en hacer cualquier otra pregunta que tengas. Además, asegúrate de informar a tu profesional de atención médica si alguien de tu familia padece la enfermedad de Wilson o presenta síntomas hepáticos, neurológicos o mentales inexplicables, ya que la enfermedad es hereditaria.
Qué esperar del médico
Es posible que el profesional de atención médica te haga varias preguntas, como las indicadas a continuación:
- ¿Los síntomas ocurren en forma continua, o aparecen y desaparecen?
- ¿Cuál es la intensidad de los síntomas?
- ¿Por cuánto tiempo has tenido estos síntomas?
- ¿Hay algo que parezca mejorar o empeorar los síntomas?
- ¿Tiene algún miembro de tu familia la enfermedad de Wilson?
Dec. 27, 2025