Descripción general
¿Qué es la fibrosis quística? Explicación de un experto de Mayo Clinic
Obtén más información de la Dra. Sarah Chalmers, neumóloga.
Hola. Soy la Dra. Sarah Chalmers, neumóloga de Mayo Clinic. En este video, hablaremos sobre los conceptos básicos de la fibrosis quística. ¿Qué es? ¿Quién puede tenerla? Cuáles son los síntomas, y cómo se puede diagnosticar y tratar. Ya sea que busques respuestas para ti o para un ser querido, estamos aquí para darte la mejor información disponible. La fibrosis quística es un trastorno que daña los pulmones, el tubo digestivo y otros órganos. Es una enfermedad hereditaria causada por un gen defectuoso que puede transmitirse de generación en generación. La fibrosis quística afecta a las células que producen moco, sudor y jugos digestivos. Estos líquidos secretados son normalmente ligeros y resbaladizos. Sin embargo, en las personas con fibrosis quística, son espesos y pegajosos. En lugar de actuar como lubricantes, estas secreciones obstruyen los tubos, los conductos y las vías respiratorias del cuerpo. Si bien no existe una cura para la fibrosis quística, las personas que padecen esta enfermedad suelen llevar una vida normal. Hay muchas herramientas y técnicas que los médicos utilizan para ayudar a controlar esta afección complicada y, gracias a la mejora de los exámenes de detección y los tratamientos, la expectativa de vida de quienes padecen la fibrosis quística es mejor que nunca.
En resumen, la fibrosis quística es un defecto genético. Un defecto de este gen cambia la forma en que una sal entra y sale de las células, lo que causa una mucosidad espesa y pegajosa en los sistemas respiratorio, digestivo y reproductivo. Es una afección hereditaria. Para padecer la fibrosis quística, un niño debe heredar una copia del gen mutado de cada padre. Si solo hereda una copia de uno de los padres, no padecerá la enfermedad. Sin embargo, será portador del gen mutado, por lo que podría transmitirlo a sus hijos en el futuro. Como la fibrosis quística es un trastorno hereditario, los antecedentes familiares determinan el riesgo. Aunque puede ocurrir en todas las razas, la fibrosis quística es más frecuente en los blancos de ascendencia de Europa del norte.
Existen dos tipos de síntomas asociados a la fibrosis quística. El primero son los síntomas respiratorios. Una mucosidad espesa y pegajosa puede obstruir las vías que transportan el aire hacia el interior y el exterior de los pulmones. Esto puede desencadenar una tos persistente que produce una mucosidad espesa, sibilancia, intolerancia al ejercicio, infecciones pulmonares reiteradas e inflamación de las fosas nasales o congestión nasal o sinusitis recurrente. El segundo son los síntomas digestivos. La misma mucosidad espesa que puede obstruir las vías respiratorias también puede obstruir los conductos que trasportan las enzimas del páncreas al intestino delgado. Esto puede causar heces con olor desagradable o grasosas, poco aumento de peso y crecimiento, obstrucción intestinal o estreñimiento crónico y grave, que puede incluir un esfuerzo frecuente de tratar de defecar. Si tú o tu hijo tienen síntomas de fibrosis quística (o si alguien de tu familia tiene fibrosis quística), habla con el médico sobre la posibilidad de realizar pruebas para detectar la enfermedad.
Dado que esta afección médica es hereditaria, es importante revisar los antecedentes familiares. Se pueden realizar pruebas genéticas para comprobar si eres portador del gen mutado que desencadena la fibrosis quística. También puede realizarse una prueba de sudor. La fibrosis quística provoca un nivel de sal en el sudor superior al normal. Los médicos examinarán el nivel de sal en el sudor para confirmar el diagnóstico.
Como esta enfermedad se transmite de padres a hijos, el cribado neonatal se realiza de forma rutinaria en todos los estados de EE. UU. El diagnóstico temprano de la fibrosis quística permite que los tratamientos se administren de manera inmediata. Desafortunadamente, no existe cura para la fibrosis quística, pero el tratamiento adecuado puede aliviar los síntomas, reducir las complicaciones y mejorar la calidad de vida. Los médicos pueden determinar la importancia de administrar ciertos medicamentos. Estos incluyen antibióticos para tratar y prevenir las infecciones pulmonares, antiinflamatorios para reducir la hinchazón de las vías respiratorias o medicamentos que diluyen la mucosidad para ayudar a expulsar la mucosidad y mejorar la función pulmonar. Los medicamentos también pueden ayudar a mejorar la función digestiva. Pueden ser ablandadores de heces, enzimas o medicamentos para reducir el ácido. Algunos medicamentos también pueden dirigirse al defecto genético que causa la fibrosis quística, y ayudan a las proteínas defectuosas a mejorar la función pulmonar y a reducir la sal en el sudor. Además de los medicamentos, las técnicas de despeje de las vías respiratorias, también llamadas “fisioterapia torácica”, pueden aliviar la obstrucción de la mucosidad y ayudar a reducir la infección y la inflamación de las vías respiratorias. Estas técnicas aflojan la mucosidad espesa en los pulmones, lo que facilita toser. En algunos casos, los médicos recurren a la cirugía para ayudar a aliviar las afecciones que pueden derivar de la fibrosis quística. Por ejemplo, la cirugía nasal y de los senos paranasales, que ayuda a respirar, o la cirugía intestinal, que ayuda a mejorar la función digestiva. En casos que ponen en riesgo la vida, se han realizado trasplantes de pulmón y de hígado. Controlar la fibrosis quística es complejo, así que considera la posibilidad de recibir tratamiento en un centro con profesionales médicos capacitados en este trastorno para evaluar y tratar la afección. También puedes preguntarle al médico sobre los ensayos clínicos. Se desarrollan constantemente tratamientos nuevos, intervenciones y pruebas para ayudar a prevenir, detectar y tratar esta afección médica.
Enterarte de que tú o alguien que conoces tiene fibrosis quística puede ser increíblemente difícil. Es normal que sientas depresión, ansiedad, enojo o miedo. Con el tiempo, encontrarás formas de afrontarlo, de encontrar apoyo y de hablar con otras personas que también estén pasando por lo mismo. Acude a tus amigos y familiares para que te ayuden a controlar el estrés y reducir la ansiedad. Busca ayuda profesional. Recuerda que las afecciones físicas conllevan una carga emocional y mental. Tómate el tiempo necesario para informarte sobre la fibrosis quística. Es un trastorno grave y complicado. No dudes en hablar con el equipo médico si tienes preguntas o inquietudes. Con los conocimientos y los tratamientos disponibles para los médicos en la actualidad, la expectativa de vida de quienes padecen la fibrosis quística es mejor que nunca. Si quieres tener aún más información acerca de la fibrosis quística, mira nuestros otros videos relacionados o visita mayoclinic.org. Te deseamos lo mejor.
La fibrosis quística es una afección hereditaria que causa daños en los pulmones, el sistema digestivo y otros órganos del cuerpo.
La fibrosis quística afecta a las células que producen mucosidad, sudor y fluidos digestivos. Estos líquidos, también llamados secreciones, suelen ser ligeros y resbaladizos, y su función es proteger los conductos internos del cuerpo y facilitar el paso a través de ellos. Pero en las personas con fibrosis quística, un gen mutado hace que las secreciones se vuelvan pegajosas y espesas. Las secreciones bloquean los conductos, especialmente en los pulmones y el páncreas.
La fibrosis quística empeora con el tiempo y requiere cuidados diarios, pero las personas con esta afección pueden asistir a clases y trabajar. Suelen tener una mejor calidad de vida que la que tenían las personas con fibrosis quística en décadas anteriores. Las mejoras en los exámenes de detección y los tratamientos se traducen en que las personas con fibrosis quística ahora pueden vivir hasta los 50 años o más, y a algunas se las diagnostica más tarde.
Fibrosis quística
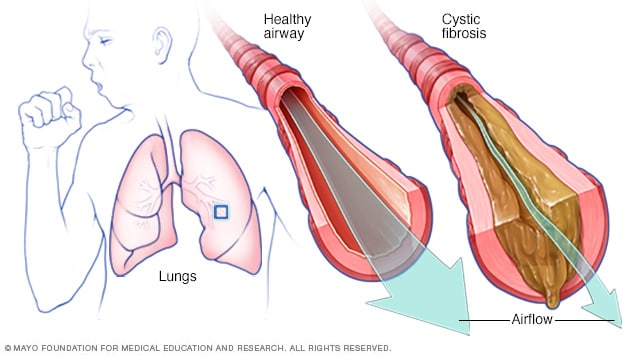
Fibrosis quística
En la presencia de fibrosis quística, las vías respiratorias se llenan con mucosidades espesas y pegajosas, lo que dificulta la respiración. La mucosidad espesa es además un caldo de cultivo ideal para las bacterias y los hongos.
Productos y servicios
Síntomas
En EE. UU., debido a los cribados neonatales, la fibrosis quística puede diagnosticarse dentro del primer mes de vida, antes de que aparezcan los síntomas. Pero las personas que nacen antes de que se realice un cribado neonatal pueden no ser diagnosticadas hasta que aparezcan los síntomas.
Los síntomas de fibrosis quística varían según los órganos afectados y la gravedad de la afección. Incluso en la misma persona, los síntomas pueden empeorar o mejorar en momentos diferentes. Es posible que algunas personas no presenten síntomas hasta la adolescencia o la edad adulta.
Las personas que no reciben un diagnóstico hasta la adultez suelen tener síntomas más leves y es más probable que presenten síntomas que no son típicos. Estos pueden incluir episodios recurrentes de inflamación del páncreas, llamada pancreatitis, infertilidad y episodios repetidos de neumonía.
Las personas con fibrosis quística tienen un nivel de sal superior al normal en su sudor. Con frecuencia, el padre y la madre pueden notar la sal cuando besan a sus hijos. La mayoría de los otros síntomas de la fibrosis quística afectan a los sistemas respiratorio y digestivo.
Síntomas respiratorios
En el caso de la fibrosis quística, lo más común es que se vean afectados los pulmones. La mucosidad espesa y pegajosa que se produce con la fibrosis quística obstruye los conductos que trasportan el aire hacia el interior y el exterior de los pulmones. Esto puede ocasionar síntomas, como los siguientes:
- Una tos que no desaparece y expulsa una mucosidad espesa.
- Un sonido chirriante al respirar llamado sibilancia.
- Capacidad limitada para realizar actividad física antes de cansarse.
- Infecciones pulmonares recurrentes.
- Conductos nasales irritados e inflamados o congestión nasal.
- Infecciones recurrentes del seno paranasal.
Síntomas digestivos
La mucosidad espesa causada por la fibrosis quística puede obstruir los conductos que llevan las enzimas digestivas del páncreas al intestino delgado. Sin estas enzimas digestivas, los intestinos no pueden asimilar ni utilizar completamente los nutrientes de los alimentos. El resultado suele ser el siguiente:
- Heces grasosas y con mal olor.
- Poco aumento de peso y crecimiento.
- Obstrucción intestinal, más frecuente en recién nacidos.
- Estreñimiento continuo o grave. Hacer mucho esfuerzo con frecuencia al intentar defecar puede hacer que parte del recto sobresalga del ano. Esto se conoce como prolapso rectal.
Cuándo debes consultar con un médico
Si tú o tu hijo tienen síntomas de fibrosis quística (o si alguien de tu familia tiene fibrosis quística) habla con tu profesional de atención médica sobre la posibilidad de realizar pruebas para detectar la afección. Programa una cita con un médico que tenga conocimientos y experiencia en el tratamiento de la fibrosis quística.
La fibrosis quística requiere un seguimiento regular con tu profesional de atención médica, al menos, cada tres meses. Llama a tu profesional de atención médica si tienes síntomas nuevos o que empeoran, como más mucosidad de lo habitual o un cambio en su color, falta de energía, pérdida de peso o estreñimiento grave.
Busca atención médica inmediata si toses sangre, tienes dolor en el pecho o dificultad para respirar, o si tienes un fuerte dolor de estómago con distensión.
Llama al 911 o al número local de servicios de emergencia o ve al Departamento de Emergencias de un hospital en los siguientes casos:
- Te cuesta recuperar el aliento o hablar.
- Los labios y las uñas se te ponen azules o grises.
- Otros notan que no estás mentalmente alerta.
Causas
Patrón hereditario autosómico recesivo
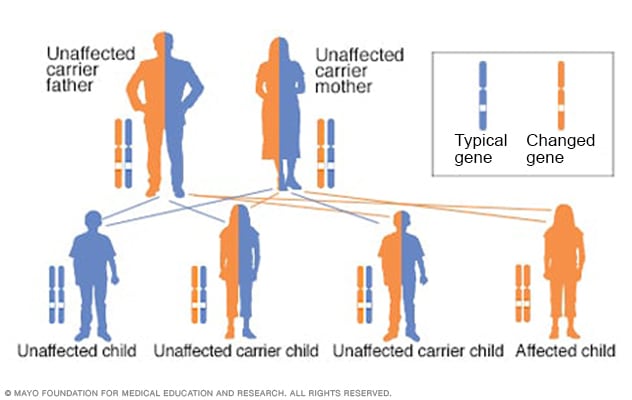
Patrón hereditario autosómico recesivo
Al tener un trastorno autosómico recesivo, heredas dos genes alterados, que a veces se denominan mutaciones. Recibes uno de cada progenitor. La salud de los padres con poca frecuencia se ve afectada porque tienen un solo gen alterado. Dos personas portadoras tienen un 25 % de probabilidades de tener un hijo no afectado, con dos genes no afectados. Tienen un 50 % de probabilidades de tener un hijo no afectado que también sea portador. Tienen un 25 % de probabilidades de tener un hijo afectado, con dos genes alterados.
En la fibrosis quística, una mutación en un gen causa problemas con la proteína que controla el movimiento de la sal y el agua dentro y fuera de las células. Este gen es el gen regulador de la conductancia transmembrana de la fibrosis quística. Afecta a las células que producen mucosidades, sudor y fluidos digestivos. Cuando la proteína reguladora de la conductancia transmembrana de la fibrosis quística no funciona como debería, el resultado es la producción de mucosidad espesa y pegajosa en los sistemas respiratorio, digestivo y reproductivo, además de una mayor cantidad de sal en el sudor.
Las mutaciones en el gen regulador de la conductancia transmembrana de la fibrosis quística que causan fibrosis quística se dividen en varios grupos según los problemas que generan. Los distintos grupos de mutaciones genéticas afectan a la cantidad de proteína reguladora de la conductancia transmembrana de la fibrosis quística que se produce y a su funcionamiento.
Para tener fibrosis quística, los niños deben heredar una copia del gen regulador de la conductancia transmembrana de la fibrosis quística mutado de cada uno de sus padres. Si los niños heredan solo una copia, no presentarán fibrosis quística. Sin embargo, serán portadores y podrían trasmitir el gen a sus propios hijos. Las personas portadoras pueden no presentar síntomas de fibrosis quística o tener solo algunos síntomas leves.
Factores de riesgo
Debido a que la fibrosis quística es una afección hereditaria, los antecedentes familiares son un factor de riesgo.
Aunque la fibrosis quística ocurre en todas las razas, es más común en la gente de piel blanca con ascendencia de Europa del Norte. Debido a que es menos común en personas de piel negra, hispanas, de Medio Oriente, nativas estadounidenses o asiáticas, el diagnóstico puede retrasarse mucho más.
Un diagnóstico tardío puede causar problemas de salud más graves. Un tratamiento temprano y eficaz puede mejorar tu calidad de vida, prevenir complicaciones y ayudarte a vivir más tiempo. Si eres una persona de color y tienes síntomas que podrían ser de fibrosis quística, habla con tu profesional de atención médica para hacerte la prueba.
Complicaciones
Las complicaciones de la fibrosis quística pueden afectar a los sistemas respiratorio, digestivo y reproductor, así como a otros órganos.
Complicaciones del sistema respiratorio
- Vías respiratorias dañadas: La fibrosis quística es una de las principales causas de la bronquiectasia, una afección de larga duración que daña las vías respiratorias. La bronquiectasia causa el ensanchamiento y la formación de cicatrices en las vías respiratorias. Esto dificulta la entrada y salida de aire de los pulmones, y la eliminación de la mucosidad de las vías respiratorias.
- Infecciones persistentes: La mucosidad espesa en los pulmones y senos paranasales crea un ambiente propicio para que bacterias y hongos vivan y proliferen. Las infecciones en los senos paranasales, la bronquitis o la neumonía son comunes y pueden ocurrir de manera repetida. También son comunes las infecciones por bacterias que no responden a los antibióticos y son difíciles de tratar.
- Protuberancias en la nariz: Debido a que el revestimiento interno de la nariz está irritado e hinchado, se pueden formar protuberancias suaves y carnosas denominadas pólipos nasales.
- Tos con sangre: La bronquiectasia puede ocurrir cerca de los vasos sanguíneos de los pulmones. La combinación de daños en las vías respiratorias y la infección puede dar lugar a tos con sangre. A menudo se trata solo de una pequeña cantidad de sangre, pero en raras ocasiones puede poner en riesgo la vida.
- Colapso pulmonar: También llamado neumotórax, esta afección ocurre cuando el aire se fuga al espacio que separa los pulmones de la pared torácica. Esto causa el colapso de una parte o la totalidad del pulmón. El colapso pulmonar es más común en adultos con fibrosis quística. El colapso pulmonar puede causar dolor repentino en el pecho y dificultad para respirar. Las personas a menudo sienten una sensación de burbujas en el pecho.
- Insuficiencia respiratoria: Con el tiempo, la fibrosis quística puede dañar el tejido pulmonar, de manera que deje de funcionar. La función pulmonar suele empeorar progresivamente y, con el tiempo, puede llegar a poner en riesgo la vida. La insuficiencia respiratoria es la causa más común de muerte por fibrosis quística.
- Episodios de empeoramiento de los síntomas: Las personas con fibrosis quística pueden pasar por períodos en los que los síntomas respiratorios empeoran más de lo habitual. Estos episodios se llaman exacerbaciones. Los síntomas pueden incluir tos con más mucosidad de lo normal y dificultad para respirar. La disminución de la energía y la pérdida de peso también son comunes durante las exacerbaciones. Las exacerbaciones se tratan con antibióticos. A veces se puede proporcionar tratamiento en casa, pero puede ser necesaria la hospitalización.
Complicaciones del sistema digestivo
- Malnutrición: La mucosidad espesa puede bloquear los conductos que trasportan las enzimas digestivas desde el páncreas hacia los intestinos. Sin estas enzimas, el cuerpo no puede asimilar ni utilizar proteínas, grasas o vitaminas liposolubles, ni tampoco puede obtener suficientes nutrientes. Esto puede causar un retraso en el crecimiento y pérdida de peso. Es común la inflamación del páncreas, una afección denominada pancreatitis.
- Diabetes: El páncreas produce insulina, que el cuerpo necesita para usar la glucosa. La fibrosis quística aumenta el riesgo de diabetes. Alrededor del 20 % de los adolescentes y hasta el 50 % de los adultos con fibrosis quística sufren de diabetes.
- Enfermedad hepática: El conducto que lleva bilis desde el hígado y la vesícula biliar hasta el intestino delgado puede bloquearse e inflamarse. Esto puede derivar en problemas hepáticos, como ictericia, enfermedad de hígado graso y cirrosis, y, a veces, cálculos biliares.
- Obstrucción intestinal: La obstrucción intestinal puede ocurrirles a las personas con fibrosis quística a cualquier edad. A veces, también puede producirse una afección en la que una sección del intestino se desliza dentro de otra sección cercana, como un telescopio plegable.
- Síndrome de obstrucción intestinal distal: El síndrome de obstrucción intestinal distal ocurre cuando se presenta una obstrucción parcial o completa en el lugar donde el intestino delgado se encuentra con el intestino grueso. El síndrome de obstrucción intestinal distal requiere tratamiento inmediato.
Complicaciones del sistema reproductor
- Infertilidad en los hombres: Casi todos los hombres con fibrosis quística son infértiles, dado que el conducto que conecta los testículos con la glándula prostática (conducto deferente) está bloqueado con mucosidad o directamente no existe. La esperma se sigue produciendo en los testículos, aunque no pueda pasar al semen generado por la glándula prostática. Ciertos tratamientos para la fertilidad y procedimientos quirúrgicos a veces hacen posible que los hombres con fibrosis quística sean padres biológicos.
- Menor fertilidad en las mujeres: Aunque las mujeres con fibrosis quística pueden ser menos fértiles que otras mujeres, es posible que conciban y tengan embarazos exitosos. Aun así, el embarazo puede empeorar los síntomas de la fibrosis quística. Habla con el profesional de atención médica sobre los riesgos.
Otras complicaciones
- Debilitamiento de los huesos: La fibrosis quística aumenta el riesgo de padecer un peligroso adelgazamiento de los huesos denominado osteoporosis. También se puede sufrir dolor en las articulaciones, artritis y dolores musculares.
- Desequilibrio de electrolitos y deshidratación: La fibrosis quística causa un sudor más salado, por lo que puede alterarse el equilibrio de minerales en la sangre. Esto aumenta el riesgo de deshidratación, especialmente con el ejercicio o cuando hace calor. Los síntomas de la deshidratación incluyen latidos cardíacos rápidos, cansancio extremo, debilidad y presión arterial baja.
- Enfermedad por reflujo gastroesofágico: El ácido del estómago fluye con frecuencia hacia el esófago, que es un conducto que conecta la boca y el estómago. Esta reacción se conoce como reflujo ácido y puede irritar la mucosa del esófago.
- Enfermedades de salud mental: Tener una enfermedad persistente que no tiene cura puede causar miedo, depresión y ansiedad.
- Mayor riesgo de cáncer en el aparato digestivo: El riesgo de cáncer de esófago, estómago, intestino delgado y grueso, hígado y páncreas es mayor en las personas con fibrosis quística. Deben comenzar a realizarse regularmente exámenes de detección para el cáncer colorrectal a los 40 años.
Prevención
Si tú o tu pareja tienen parientes cercanos con fibrosis quística, ambos pueden elegir hacerse pruebas genéticas antes de tener hijos. La prueba, que se realiza en un laboratorio con una muestra de sangre, puede ayudar a determinar el riesgo de tener un hijo con fibrosis quística.
Si ya estás embarazada y la prueba genética muestra que tu bebé puede estar en riesgo de padecer fibrosis quística, el profesional de atención médica puede realizar otras pruebas a tu hijo por nacer.
Las pruebas genéticas no son para todos. Antes de que decidas hacerte la prueba, habla con un consejero genético sobre el impacto en la salud mental que pueden tener los resultados de la prueba.