نظرة عامة
يؤدي داء هنتنغتون إلى تحلل الخلايا العصبية في الدماغ بمرور الوقت. يؤثر الداء في حركات الشخص المصاب وقدرات التفكير لديه وصحته العقلية.
داء هنتنغتون مرض نادر. وغالبًا ما ينتقل عبر وراثة جين متغير من أحد الوالدين.
يمكن أن تظهر أعراض داء هنتنغتون في أي وقت، لكن غالبًا ما يبدأ ظهورها خلال الثلاثينيات أو الأربعينيات من العمر. في حال الإصابة بالمرض قبل عمر 20 عامًا، فتُسمى هذه الحالة داء هنتنغتون في اليافعين. وعند الإصابة بداء هنتنغتون مبكرًا، يمكن أن تختلف الأعراض وقد يتقدّم المرض بشكل أسرع.
تتوفر الأدوية للمساعدة على إدارة أعراض داء هنتنغتون. ولكن لا يمكن للعلاجات أن تمنع التدهور الجسدي والعقلي والسلوكي الناجم عن المرض.
المنتجات والخدمات
الأعراض
عادةً ما يسبب داء هنتنغتون حدوث اضطرابات حركية. كما أنه يسبب حالات صحية عقلية ومشكلات في التفكير والتخطيط. يمكن أن تسبب هذه الحالات مجموعة كبيرة من الأعراض. وتختلف الأعراض التي تبدأ في الظهور أولاً اختلافًا كبيرًا من شخص لآخر. وتظهر بعض الأعراض بشكل أكثر شدة أو يكون لها تأثير كبير في القدرة الوظيفية. ومع ذلك من الممكن أن تتغير الأعراض في شدتها خلال فترة المرض.
اضطرابات الحركة
قد تسبب اضطرابات الحركية المرتبطة بداء هنتنغتون حركات لا إرادية، وتسمى الرُّقاص. الرُّقاص هي حركات لا إرادية تؤثر في كافة عضلات الجسم، وتحديدًا الذراعين والساقين، والوجه واللسان. كما يمكن أن تؤثر في القدرة على القيام بحركات إرادية. تشمل الأعراض ما يأتي:
- حركات اهتزازية أو متلوية لا إرادية.
- تصلب العضلات أو تقلصها.
- حركات بطيئة أو غير معتادة للعين.
- خلل في المشي ووضعية الجسم والتوازن.
- صعوبة في التحدث أو البلع.
قد لا يتمكن الأشخاص المصابون بداء هنتنغتون أيضًا من التحكم في الحركات الإرادية. ويمكن أن يكون لهذا تأثير أكبر من الحركات اللاإرادية التي يسببها المرض. حيث قد تؤثر مشكلة الحركات الإرادية في قدرة الشخص على العمل وأداء الأنشطة اليومية والتواصل والحفاظ على استقلاليته.
الأمراض المعرفية
يؤدي داء هنتنغتون في الغالب إلى حدوث مشكلات في المهارات المعرفية. وقد تشمل هذه الأعراض ما يأتي:
- صعوبة في تنظيم المهام أو تحديد أولويتها أو التركيز عليها.
- قلة المرونة أو التكرار المستمر لفكرة أو سلوك أو فعل، ويُعرف هذا باسم المواظبة.
- عدم التحكم في الاندفاع الذي يمكن أن يؤدي إلى نوبات من الانفعال، والتصرف من دون تفكير، وتعدد العلاقات الجنسية.
- قلة الوعي بسلوكيات الفرد وقدراته.
- البطء في معالجة الأفكار أو "العثور" على الكلمات.
- صعوبة اكتساب معلومات جديدة.
مشكلات الصحة العقلية
الاكتئاب أكثر حالات الصحة العقلية المرتبطة بداء هنتنغتون شيوعًا. وهذا ليس مجرد رد فعل لتشخيص الإصابة بداء هنتنغتون. لكن يبدو أن الاكتئاب يحدث بسبب تضرر في الدماغ وتغيرات في وظائف الدماغ. تشمل الأعراض ما يأتي:
- سهولة الاستثارة أو الحزن أو اللامبالاة.
- الانسحاب الاجتماعي.
- صعوبة في النوم.
- التعب وفقدان الطاقة.
- أفكار عن الموت أو الوفاة أو الانتحار.
تشمل حالات الصحة العقلية الشائعة الأخرى ما يأتي:
- اضطراب الوسواس القهري، حالة تتسم بوجود الأفكار المقتحمة التي يتكرر حدوثها والسلوكيات المتكررة مرارًا وتكرارًا.
- الهوس، يمكن أن يسبب مزاجًا حادًا وفرطًا في النشاط وسلوكًا اندفاعيًا وتقدير الذات بشكل مبالغ فيه.
- الاضطراب ثنائي القطب، حالة تتناوب فيها نوبات الاكتئاب والهوس.
يشيع فقدان الوزن أيضًا لدى الأشخاص المصابين بداء هنتنغتون وخصوصًا مع تفاقم المرض.
أعراض مرض هنتنغتون مجهول السبب
يظهر داء هنتنغتون ويتقدم بطريقة مختلفة قليلاً لدى الشباب عن الطريقة التي يظهر ويتقدم بها لدى البالغين. وتشمل الأعراض التي تظهر غالبًا في المراحل المبكرة من مسار المرض ما يأتي:
التغيرات السلوكية
- صعوبة الانتباه.
- انخفاض مفاجئ في الأداء المدرسي العام.
- المشكلات السُّلوكية، مثل سلوك عدواني أو تخريبي.
التغيرات البدنية
- عضلات متقلصة وصلبة تؤثر في المشي خصوصًا عند الأطفال الصغار.
- حركات خفيفة لا يمكن السيطرة عليها، تعرف باسم الرُّعاش.
- حالات السقوط المتكرِّرة أو التخبط أثناء الحركة.
- نوبات الصرع.
متى تزور الطبيب
استشِر اختصاصي الرعاية الصحية إذا لاحظت تغيرات في حركاتك أو حالتك العاطفية أو قدرتك العقلية. يمكن أيضًا أن تنتج أعراض داء هنتنغتون عن عدد من الأمراض المختلفة. لذا، من المهم تشخيص الحالة بشكل سريع وشامل.
الأسباب
نمط وراثي صبغي جسدي سائد
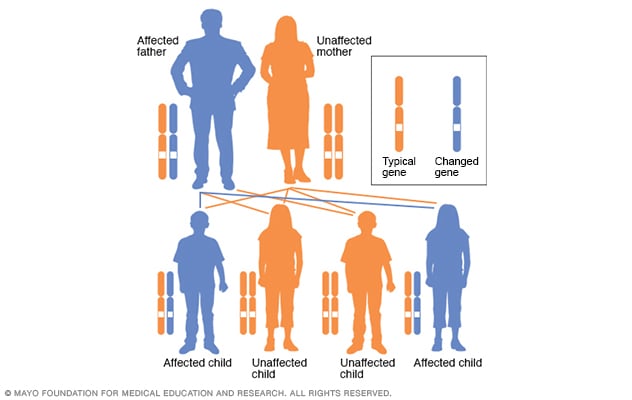
نمط وراثي صبغي جسدي سائد
في حالات الاضطراب الصبغي الجسدي السائد، يكون الجين المتحور جينًا سائدًا. ويوجد في أحد الصبغيات (الكروموسومات) اللاجنسية التي تُسمى بالصبغيات الجسدية. ولا يحتاج الأمر سوى جينٍ متحور واحد فقط للإصابة بهذا النوع من الحالات. ويكون لدى الشخص المصاب بحالة الاضطراب الصبغي الجسدي السائد، وهو الأب في هذه الحالة، احتمال بنسبة 50% لإنجاب طفل مصاب بجين متحور واحد. وبالتالي، تتساوى احتمالات إنجاب طفل مصاب أو غير مصاب بهذا الاضطراب.
ينتج داء هنتنغتون عن اختلاف في جين واحد ينتقل من أحد الوالدين. يتبع داء هنتنغتون نمطًا وراثيًا صبغيًا سائدًا. وهذا يعني أن الشخص يحتاج إلى نسخة واحدة فقط من الجين غير الطبيعي لحدوث اضطراب.
يرث الإنسان نسختين من كل جين، واحدة من كلا أبويه، وذلك باستثناء الجينات الموجودة في الصبغيات (الكروموسومات) الجنسية. يمكن لأحد الوالدين الذي يحمل جينًا غير طبيعي أن ينقل إما النسخة غير الطبيعية من ذلك الجين أو النسخة السليمة منه. وعندئذ يكون هناك احتمال بنسبة 50% أن يرث كل طفل في العائلة الجين المسبب للإصابة بالحالة الوراثية.
عوامل الخطورة
الأشخاص الذين لديهم أحد الوالدين مصاب بداء هنتنغتون معرضون لخطر الإصابة بالمرض. الأطفال الذين لديهم أحد الوالدين مصاب بداء هنتنغتون لديهم فرصة بنسبة 50% للإصابة بالتغير الجيني الذي يسبب مرض هنتنغتون.
المضاعفات
بعد بدء ظهور داء هنتنغتون، تتدهور القدرات الوظيفية تدريجيًا بمرور الوقت لدى الشخص المصاب. وتختلف سرعة تدهور المرض والمدة التي يستغرقها من شخص لآخر. فتتراوح المدة منذ بداية ظهور الأعراض حتى الوفاة بين 10 و30 عامًا. ويؤدي داء هنتنغتون في اليافعين عادةً إلى الوفاة في غضون 10 إلى 16 عامًا بعد بدء ظهور الأعراض.
قد يؤدي الاكتئاب الذي يرافق داء هنتنغتون إلى زيادة خطر الانتحار. تشير بعض الأبحاث إلى أن خطر الانتحار يكون أكبر قبل التشخيص وكذلك عند فقدان الشخص استقلاليته.
في نهاية المطاف، يحتاج المصاب بداء هنتنغتون إلى المساعدة على أداء جميع أنشطة حياته اليومية ورعايته. وفي مرحلة متأخرة من المرض، سيكون الشخص على الأرجح حبيس الفراش ولن يكون قادرًا على الكلام. وبشكل عام، يكون الشخص المصاب بداء هنتنغتون قادرًا على فهم اللغة والتعرف على أفراد العائلة والأصدقاء، إلا أن البعض لا يكون قادرًا على التعرف على أفراد العائلة.
تتضمن الأسباب الشائعة للوفاة:
- التهاب الرئة أو العدوى الأخرى.
- الإصابات الناجمة عن السقوط.
- المضاعفات المتعلقة بصعوبة في البلع.
الوقاية
يشعر الأشخاص الذين لديهم سيرة مرضية عائلية معروفة للإصابة بداء هنتنغتون بالقلق بشأن احتمالية نقلهم لجين هنتنغتون إلى أطفالهم. من الممكن أن يلجأوا إلى إجراء فحص جيني والتفكير بخيارات تنظيم الأسرة.
إذا كان أحد الوالدين المعرضين للخطر يفكر في إجراء اختبار الجينات، فقد يكون من المفيد مقابلة استشاري في الأمراض الوراثية. سيناقش استشاري الأمراض الوراثية المخاطر المحتملة لنتيجة الاختبار الإيجابية، والتي من شأنها أن تشير إلى احتمالية إصابة أحد الوالدين بالمرض. كما سيحتاج الأزواج إلى التفكير في خيارات إضافية بشأن إنجاب الأطفال أو التفكير في بدائل. وقد يقرران إجراء اختبار الجين أثناء فترة الحمل أو الإخصاب في المختبر بالاستعانة بحيوانات منوية أو بويضات من أحد المتبرعين.
هناك خيار آخر للأزواج وهو الإخصاب في المختبر والتشخيص الوراثي السابق لانغراس البويضة. في هذه العملية، تُنقل البويضات من الـمِبيَضين وتُخصَّب بالحيوانات المنوية للأب داخل المختبر. يُجرى اختبار للأجنة بحثًا عن وجود جين هنتنغتون. وتُزرَع الأجنة التي تثبت سلبية جين هنتنغتون في رحم الأم.
الإخصاب المخبري
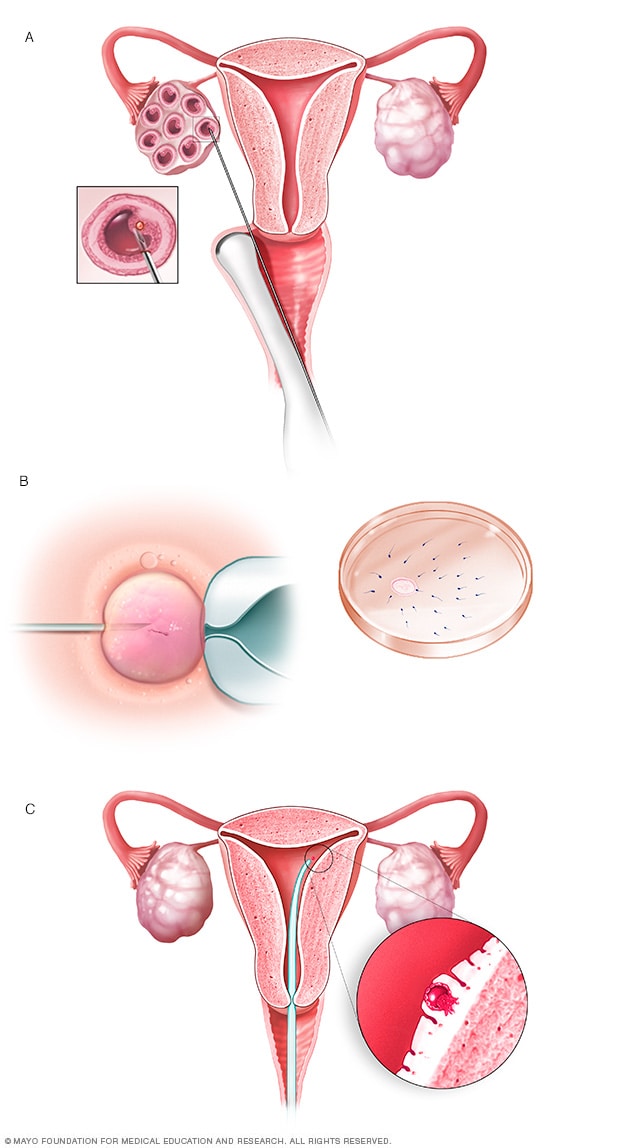
الإخصاب المخبري
أثناء الإخصاب المخبري، تُزال البويضات من أكياس تُسمى الجريبات داخل أحد المبيضين (أ).
تُخصَّب البويضة من خلال حقن حيوان منوي واحد في البويضة أو خلطها بالحيوان المنوي في طبق بِترِي (ب).
تُنقل البويضة المخصبة، التي يطلق عليها الجنين، إلى الرحم (جـ).