نظرة عامة
نقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة هو اضطراب وراثي يمنع الجسم من تكسير دهون معينة وتحويلها إلى طاقة. يتضمن الأيض العمليات التي يستخدمها الجسم لإنتاج الطاقة. وقد يُسبب نقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة مشكلات في الأيض.
قد يؤدي نقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة في حال عدم علاجه إلى نقص شديد في الطاقة والإرهاق ومرض الكبد والغيبوبة وغير ذلك من المشكلات الصحية الخطيرة. وقد يؤدي أيضًا إلى انخفاض مستوى السكر في الدم إلى مستويات خطيرة؛ ما يُطلق عليه نقص سكر الدم. تجدر الإشارة إلى الوقاية والعلاج الفوري عنصران مهمان بغض النظر عن مستوى السكر في الدم. إذا كنت مصابًا بنقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة، فيمكن أن تحدث نوبة مفاجئة، تسمَّى أزمة الأيض، بسبب الأمراض الشائعة أو الحمى الشديدة أو مشكلات المعدة أو عدم تناول الطعام لفترة طويلة، في ما يُعرف باسم الصيام لفترات طويلة.
حالة نقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة هي حالة توجد منذ الولادة وتستمر مدى الحياة. وتُجري جميع الولايات في الولايات المتحدة فحص نقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة عند الولادة كجزء من فحص حديثي الولادة. وتوفر العديد من الدول الأخرى فحصًا روتينيًا لنقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة لدى حديثي الولادة. ويمكن السيطرة جيدًا على اضطراب نقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة في حال تشخيصه وعلاجه مبكرًا باتباع نظام غذائي ونمط حياة صحي.
الأعراض
عادةً يُلاحَظ نقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة (MCAD) لأول مرة عند الرُّضَّع والأطفال الصغار. وفي حالات نادرة، لا يُكتشف الاضطراب إلا عند البلوغ.
يمكن أن تختلف أعراض نقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة من مريض إلى آخر. وقد تشمل ما يلي:
- القيء.
- انخفاض الطاقة أو انعدامها.
- الشعور بالضعف.
- انخفاض مستوى السكر في الدم.
يمكن أن تحدث نوبة شديدة مفاجئة، تسمى أزمة الأيض، بسبب:
- البقاء مدة طويلة من دون تناول الطعام.
- تفويت وجبات.
- العَدوى الشائعة.
- الحُمى الشديدة.
- مشكلات مستمرة في المعدة والهضم قد تسبب القيء والإسهال.
- التمارين الرياضية المكثفة.
متى تجب زيارة الطبيب
يخضع حديثو الولادة في الولايات المتحدة والكثير من البلدان الأخرى لبرامج فحص للتأكد من عدم إصابتهم بنقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة (MCAD). وقد تُحالون بعد التقييم الأول إلى اختصاصي مُدرّب على تقييم نقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة وعلاجه. قد تُحالون كذلك إلى أعضاء آخرين في فريق الرعاية الصحية، مثل اختصاصي النُّظم الغذائية المسجَّل.
اتصل بفريق الرعاية الصحية إذا شُخصت بالإصابة بنقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة، وإذا كنت مصابًا بحُمى شديدة أو انعدام الشهية أو أعراض في المعدة أو الجهاز الهضمي أو خضعت لإجراء طبي مخطط له يتطلب الصيام.
الأسباب
نمط وراثي صبغي جسدي متنحٍ
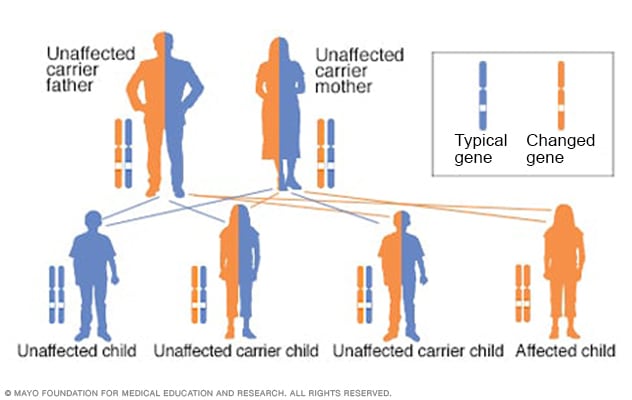
نمط وراثي صبغي جسدي متنحٍ
تحدث الإصابة بالاضطراب الصبغي الجسدي المتنحي عندما يرث الشخص جينَين من الجينات المتغيرة، وتُسمى أحيانًا طفرات. فيرث الشخص جينًا من الأب وجينًا من الأم. ونادرًا ما تتأثر صحتهما، لأن كلًّا منهما يحمل جينًا متغيرًا واحدًا. ويكون لدى الشخصين الحاملين للصفة الوراثية احتمال بنسبة 25% في إنجاب طفل غير مصاب لديه جينان طبيعيان، واحتمال بنسبة 50% في إنجاب طفل غير مصاب يكون حاملاً أيضًا للصفة الوراثية، واحتمال بنسبة 25% في إنجاب طفل مصاب لديه جينان من الجينات المتغيرة.
في حال عدم احتواء الجسم على ما يكفي من إنزيم نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة، فإنه لا يستطيع تكسير بعض الدهون التي تُسمَّى الأحماض الدهنية متوسطة السلسلة وتحويلها إلى طاقة. ويؤدي ذلك إلى انخفاض الطاقة وانخفاض مستوى السكر في الدم. وقد تتراكم أيضًا الأحماض الدهنية في أنسجة الجسم وتسبب تلفًا.
يحدث نقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة بسبب تغير في جين ACADM. وتنتقل هذه الحالة بالوراثة من الوالدَين بنمط صبغي جسدي متنحٍ. ويعني يعني هذا أن الوالدين حاملان للصفة الوراثية -أي أن كلاً منهما يحمل جينًا واحدًا متغيرًا وجينًا واحدًا غير متغير- لكن لا تظهر عليهما أعراض الحالة. يرث الطفل المصاب بنقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة نسختين من الجين المتغير؛ واحدة من الأب وواحدة من الأم.
إذا ورثت جينًا واحدًا متغيرًا فقط، فلن تصاب بنقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة. ولكن عندما يكون لديك جين واحد متغير، تصبح حاملاً لهذا الجين المتغير ويمكن أن تنقله إلى أطفالك. ومع ذلك، فلن يصابوا بهذه الحالة ما لم يرثوا جينًا متغيرًا أيضًا من الأم.
عوامل الخطورة
يكون الطفل معرضًا لخطر الإصابة بنقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة إذا كان كلا الوالدين حاملين لجين معروف بأنه يسببها. يرث الطفل لاحقًا نسختين من الجين — واحدة من الأب وواحدة من الأم. لا يصاب الأطفال الذين يرثون نسخة واحدة فقط من الجين المصاب من أحد الوالدين عادةً بنقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة لكنهم سيكونون حاملين للجين.
المضاعفات
إذا تُركت أزمة التمثيل الغذائي الناجمة عن نقص نازعة هيدروجين أسيل التميم الإنزيمي A متوسط السلسلة دون علاج، فقد تؤدي إلى:
- نوبات صرع.
- مشكلات في الكبد.
- تضرر الدماغ.
- غيبوبة.
- وفاة مفاجئة.