Oct. 22, 2019
Characteristics of three patients with FPLD
Familial partial lipodystrophy (FPLD) is a heterogeneous group of rare inherited disorders characterized by varying degrees of fat loss and metabolic abnormalities. The severity of metabolic derangements correlates with the extent of fat loss, thus highlighting the critical role of adipose tissue in glucose and lipid metabolism. The following vignettes highlight the genotypic and phenotypic diversity of FPLD and associated metabolic abnormalities and comorbidities, and discuss standard management approaches for patients with these disorders.
Case 1: Type 2 FPLD (Dunnigan variety)
A 40-year-old Asian Indian female belonging to a large pedigree harboring the LMNA R482W mutation presented to the metabolism clinic at Mayo Clinic in Rochester, Minnesota. She had normal fat distribution as a child, but noted onset of fat loss from the limbs at age 10. Menarche occurred at 13, but cycles were irregular. At age 26, the patient was diagnosed with type 2 diabetes mellitus (T2DM) and had developed excess fat in her face, neck and back. Other metabolic complications included hypertriglyceridemia, fatty liver disease and polycystic ovary syndrome (PCOS). Her physical exam revealed a BMI of 19.42 kg/m2, absent fat in the extremities with muscular prominence, excess fat over the face and posterior neck, as well as acanthosis.
The patient's mother, maternal aunt and several other maternal relatives had similar histories and phenotypes, including premature coronary artery disease in two family members. Notably, the patient's 12-year-old daughter also had clinical features of type 2 FPLD.
Case 2: Type 1 FPLD (Köbberling variety)
Coronal CT images of cases 2 and 3
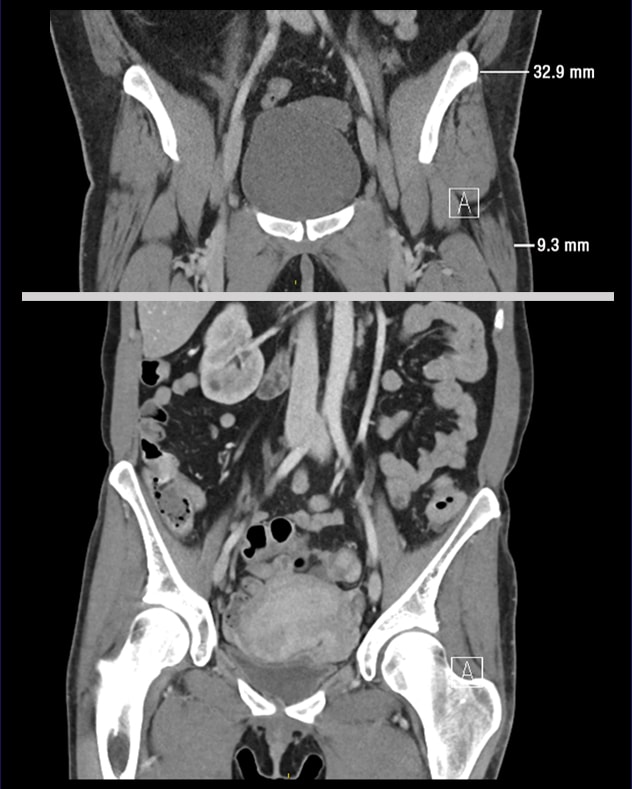
Coronal CT images of cases 2 and 3
Coronal CT images of case 2 with type 1 FPLD (top) and case 3 with LMNA mutation-associated generalized lipodystrophy (bottom). Note the excess adiposity in the trunk and loss in the hips and legs of case 2 compared with the generalized loss of fat from trunk, hips and legs of case 3.
Sagittal CT images of cases 2 and 3
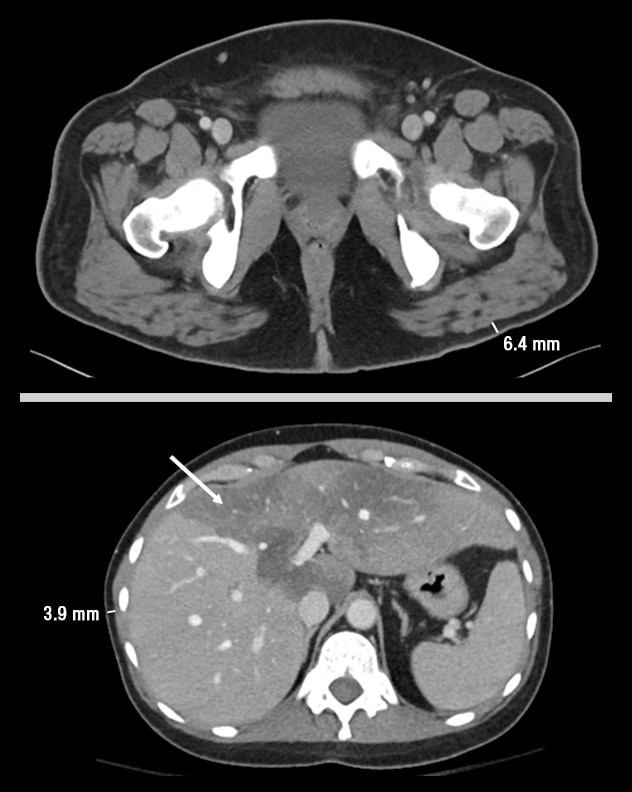
Sagittal CT images of cases 2 and 3
Sagittal CT images of case 2 with type 1 FPLD (top) and case 3 with LMNA mutation-associated generalized lipodystrophy (bottom). Note the fat loss in the gluteal region of case 2. Note the generalized loss of truncal fat and hepatomegaly with steatosis (arrow) in case 3.
A 31-year-old Caucasian female was evaluated for hypertriglyceridemia and T2DM. She had normal fat distribution until puberty, at which point she experienced fat loss in her extremities and fat excess in her abdomen, neck and face. Metabolic complications included PCOS, obesity (BMI 38.5 kg/m2), fatty liver disease, hypertriglyceridemia complicated by pancreatitis, and T2DM requiring high doses of insulin. Her examination revealed excess adiposity in the face, neck and trunk; acanthosis; and fat loss with muscular prominence of the extremities and gluteal area.
Her mother, sister, maternal grandmother, and several maternal aunts and uncles had similar physical features. One maternal uncle had a myocardial infarction at age 40.
Case 3: LMNA-associated generalized lipodystrophy
A 44-year-old female, also belonging to a large pedigree with lipodystrophy, began losing fat from the face and limbs in her late 20s, with relative sparing of the abdomen. At that time, she also developed gray hair, skin wrinkling and hearing impairment. In subsequent years she was diagnosed with T2DM, hypertriglyceridemia, fatty liver disease, nonischemic cardiomyopathy, focal segmental glomerulonephritis, and glottis squamous cell carcinoma. The patient's BMI was 16 kg/m2. Her physical exam revealed generalized fat loss involving all extremities, palms, soles and face. She also had progeroid features, a prominent systolic murmur and hepatomegaly.
Genetic testing revealed a mutation involving the LMNA gene (c.1045C>T, p.R349W).
Her mother, maternal uncle and two siblings had similar physical features with onset in their 20s, and all had developed cardiomyopathy.
Discussion
As noted in research published in The Journal of Clinical Endocrinology & Metabolism in 2011, FPLD is a rare autosomal dominant disorder characterized by loss of subcutaneous fat from the extremities with sparing of the face, neck and variably the trunk. Onset occurs during childhood, puberty or later. To date, seven subtypes have been described, six of which are related to mutations in genes involved in adipose metabolism and development: LMNA, PPARG, AKT2, CIDEC, PLIN1 and LIPE, though the exact mechanism by which these mutations lead to fat loss remains unclear. In these disorders both fat loss and subsequent caloric excess result in severe insulin resistance, hepatic steatosis, hypertriglyceridemia, low HDL, PCOS and cardiovascular disease.
Type 2 FPLD (Dunnigan variety) is the most well-described subtype. It is caused by a mutation in the LMNA gene. As demonstrated by case 1, the typical phenotype involves fat loss from the extremities with sparing of the face and neck.
Case 2 represents type 1 FPLD (Köbberling variety), the genetic basis of which has not been identified. This phenotype involves fat loss from the extremities and gluteal regions, often with excess fat over the trunk, face and neck.
Case 3 represents a rare phenotype of FPLD. It also involves a mutation of the LMNA gene (c.1045C>T, p.R349W), but instead of causing fat loss from the extremities with sparing of the face and neck, it causes generalized lipodystrophy. In addition to the common metabolic abnormalities of FPLD, this variety is associated with progeroid features, focal segmental glomerulonephritis, cardiomyopathy and cardiac conduction defects.
It is important to recognize FPLD and its different subtypes in patients being evaluated for insulin resistance, hyperlipidemia, PCOS, nonalcoholic steatohepatitis and Cushing syndrome. Skinfold measurement of the anterior thigh and other appendicular and truncal sites can be helpful, although distinct cutoffs have not been standardized.
Management for the metabolic complications of FPLD centers on dietary restriction of saturated fats and refined carbohydrates, as well as traditional glucose and lipid-lowering therapies. Recognition of the different FPLD subtypes will aid in screening for appropriate metabolic complications and comorbidities.
For more information
Garg A. Lipodystrophies: Genetic and acquired body fat disorders. The Journal of Clinical Endocrinology & Metabolism. 2011;96:3313.