Descripción general
La enfermedad de Gaucher es una afección poco común y hereditaria que deriva en acumulación de una sustancia grasa en los tejidos. La acumulación de esta sustancia daña y afecta el funcionamiento de tejidos y órganos. Los órganos más afectados son el bazo, el hígado y los huesos.
Existen tres tipos generales de enfermedad de Gaucher. Más del 90 % de los casos son del tipo 1.
Los tratamientos principales descomponen la sustancia grasa o limitan su producción. Los profesionales hacen pruebas regulares para vigilar los tratamientos y buscar señales de daño en los tejidos.
La enfermedad de Gaucher tipo 1 es más común en personas con ascendencia judía del este y centro de Europa, quienes se conocen como judíos asquenazíes.
Productos y servicios
Síntomas
Algunos síntomas son comunes a todos los tipos de enfermedad de Gaucher, pero cada tipo tiene diferencias importantes.
Tipo 1
La gravedad de los síntomas puede variar. En algunos casos, un profesional de atención médica puede detectar signos de la enfermedad antes de que aparezcan los síntomas. Los síntomas suelen aparecer en la niñez o adolescencia, pero pueden comenzar a cualquier edad. Algunos de los signos y síntomas de la enfermedad de Gaucher tipo 1 son los siguientes:
- Órganos agrandados. El bazo se agranda mucho. El hígado también puede agrandarse. Los efectos pueden incluir los siguientes:
- Moretones o sangrado fáciles debido a que las plaquetas de la sangre quedan atrapadas en el bazo.
- Daño tisular o formación de cicatrices en el bazo o el hígado.
- Dolor o molestias en el abdomen.
- Afecciones de los huesos Los huesos debilitados e irregulares pueden causar lo siguiente:
- Dolor en los huesos.
- Fracturas óseas, incluso sin una lesión aparente.
- Trastornos de la médula ósea. La médula ósea puede no producir suficientes células sanguíneas sanas, lo que causa lo siguiente:
- Baja producción de plaquetas, que deriva en facilidad para formar moretones o sangrado.
- Baja producción de glóbulos rojos, que causa fatiga.
- Enfermedad pulmonar. Con menos frecuencia, la enfermedad puede afectar la respiración debido a lo siguiente:
- Daño a los tejidos pulmonares.
- Restricciones en los pulmones ocasionadas por un agrandamiento del bazo o del hígado.
- Presión arterial alta en los pulmones.
- Trastornos de crecimiento. Algunos niños con la enfermedad de Gaucher tipo 1 presentan lo siguiente:
- Retraso en el crecimiento o crecimiento limitado.
- Retraso en la pubertad.
Tipo 2
La enfermedad de Gaucher tipo 2 es la menos común. Causa una rápida pérdida de células en el tallo cerebral. Esta parte del cerebro regula la respiración, el control muscular y otras funciones importantes.
Los síntomas aparecen durante los primeros meses de vida. La muerte suele ocurrir en el plazo de dos años.
Tipo 3
La enfermedad de Gaucher tipo 3 causa síntomas similares a los de la enfermedad de Gaucher tipo 1. También causa algunos trastornos del sistema nervioso y la pérdida progresiva de neuronas cerebrales. Los síntomas comienzan durante la niñez. Entre estos, se incluyen los siguientes:
- Trastornos del movimiento ocular.
- Espasmos musculares.
- Convulsiones.
- Pérdida del control muscular en la adolescencia o adultez temprana.
- Demencia en la adolescencia o adultez temprana.
Cuándo debes consultar a un médico
Si tú o tu hijo tienen los síntomas relacionados con la enfermedad de Gaucher, pide una cita con un profesional de atención médica.
Causas
Patrón hereditario autosómico recesivo
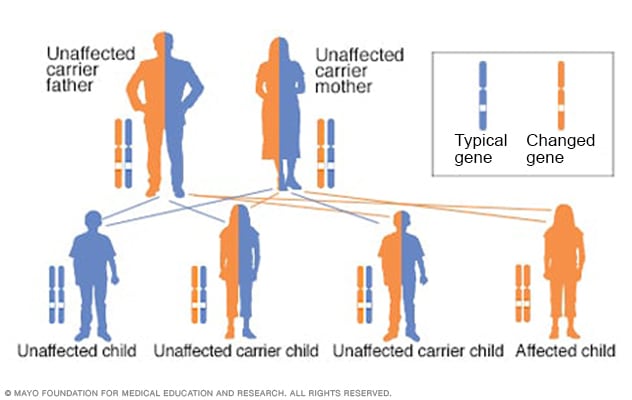
Patrón hereditario autosómico recesivo
Al tener un trastorno autosómico recesivo, heredas dos genes alterados, que a veces se denominan mutaciones. Recibes uno de cada progenitor. La salud de los padres con poca frecuencia se ve afectada porque tienen un solo gen alterado. Dos personas portadoras tienen un 25 % de probabilidades de tener un hijo no afectado, con dos genes no afectados. Tienen un 50 % de probabilidades de tener un hijo no afectado que también sea portador. Tienen un 25 % de probabilidades de tener un hijo afectado, con dos genes alterados.
Una sustancia grasa, llamada glucocerebrósido, forma parte de la pared de la célula y cumple una función importante en la actividad celular. Cuando una célula muere, una enzima específica descompone esta sustancia grasa.
En la enfermedad de Gaucher, el gen responsable de producir esta enzima presenta una mutación, es decir que tiene un cambio anormal. Las sustancias grasas no se descomponen. En su lugar, se acumulan en los glóbulos blancos que se encargan de eliminar los desechos de las células muertas.
Estos glóbulos blancos anormales, llamados células de Gaucher, dañan los tejidos.
Patrón de herencia
La enfermedad de Gaucher se transmite a un niño cuando el padre y la madre tienen una mutación genética relacionada con esta enfermedad. El niño recibe un gen mutado del padre y de la madre. Este patrón de herencia se llama autosómico recesivo.
Los niños que heredan solo un gen mutado no presentan la enfermedad, pero pueden transmitir la mutación.
Factores de riesgo
Las personas con ascendencia judía del este y centro de Europa, conocidas como judíos asquenazíes, tienen un riesgo más alto que la población general de presentar la enfermedad de Gaucher tipo 1.
Complicaciones
Algunas de las complicaciones de la enfermedad de Gaucher tipo 1 son las siguientes:
- Riesgo de padecer enfermedad de Parkinson. Las personas con enfermedad de Gaucher tipo 1 tienen un mayor riesgo de presentar la enfermedad de Parkinson en comparación con la población general. Quienes tienen una sola copia de la mutación genética relacionada con la enfermedad de Gaucher también presentan un riesgo más alto. Además, existe más riesgo de padecer un trastorno relacionado llamado demencia con cuerpos de Lewy.
- Riesgo de cáncer. Las personas con enfermedad de Gaucher tipo 1 también tienen más riesgo de presentar cáncer de médula ósea y de la sangre.
- Complicaciones en el embarazo. Los síntomas de la enfermedad de Gaucher pueden empeorar durante el embarazo. También existe más riesgo de sangrado después del parto.
Oct. 25, 2025