Feb. 14, 2020
Hereditary causes, due to defects in certain genes, account for up to 10% of all colorectal cancers (CRCs). These high-risk hereditary predisposition syndromes have been associated with a significantly increased lifetime risk of cancer, with some approaching 100%. In addition, up to 20% of patients with CRC have common familial cancer, meaning there is a strong family history of CRC but no one genetic defect has been identified.
In an article published in Mayo Clinic Proceedings in 2019, Niloy Jewel (Jewel) Samadder, M.D., and co-authors present a primer on the diagnosis and management of these hereditary conditions and the gastrointestinal cancers with which they are associated. Dr. Samadder is a gastroenterologist and hepatologist specializing in inherited cancers at Mayo Clinic's campus in Arizona.
In this article, Dr. Samadder shares information about Lynch syndrome (LS), familial adenomatous polyposis (FAP) and attenuated FAP (AFAP) to provide clinicians with tools to understand the genetic bases of these conditions and the appropriate diagnosis and management.
Lynch syndrome (LS)
Overview and clinical presentation
Hereditary nonpolyposis CRC, also known as LS, is the most common hereditary CRC predisposition syndrome and accounts for 2% to 4% of all colorectal neoplasias. Individuals with LS have an elevated lifetime risk of CRC (80%) and endometrial cancer (60%).
In addition to colorectal and endometrial cancers, individuals with LS have a predisposition for these extracolonic cancers: gastric, ovarian, breast, liver and bile duct, genitourinary, pancreatic, small bowel, brain, sebaceous gland adenomas, and keratoacanthomas. The lifetime risk of each of these cancers varies and is affected by the individual's age, sex and the specific mismatch repair (MMR) germline mutation involved.
Genetics and diagnosis
LS is an autosomal dominant disorder caused by a germline mutation in one of several DNA MMR genes on chromosome 3p21 that are responsible for post-replicative proofreading — MLH1, MSH2, MSH6, PMS2 and EPCAM.
"The accelerated rate at which these cancers develop and the associated risk of multiple malignancies make early diagnosis critical," explains Dr. Samadder. In addition to a review of family and personal cancer history, the Amsterdam and revised Bethesda criteria can help identify patients carrying mutations.
Tumor testing for LS can include microsatellite instability (MSI) status testing and immunohistochemical (IHC) analysis to assess for MMR protein expression. Current guidelines recommend these tests for every CRC tumor specimen to identify LS-related cancers. If multigene testing is performed, it should be accompanied by professional pretest and post-test counseling.
Colorectal cancer surveillance and management
The National Comprehensive Cancer Network and others recommend performing surveillance colonoscopy every one to two years in confirmed LS mutation carriers, beginning at age 20 to 25, or two to five years before the age at which the youngest family member was diagnosed, whichever comes first.
Patients with LS who are diagnosed with CRC may benefit from a completion colectomy with ileorectal anastomosis to eliminate future risk of CRC. A segmental resection may be considered if postoperative surveillance can be performed every one to two years.
Extracolonic cancer surveillance and management recommendations
Currently there are no universal guidelines to guide screening for extracolonic cancers associated with LS. A few general recommendations for each cancer are listed below.
Endometrial cancer: Patient education about symptoms and instructions to report symptoms; annual endometrial biopsy can also be considered.
Ovarian cancer: Transvaginal ultrasonography and serum cancer antigen 125 measurement can be considered, although data about their efficacy are limited. To reduce the risk of both endometrial and ovarian cancers, prophylactic total abdominal hysterectomy and bilateral salpingo-oophorectomy are recommended in women beyond the childbearing years.
Gastric and duodenal cancers: Baseline esophagogastroduodenoscopy, extended to the distal duodenum, by age 30 to 35 years; gastric biopsies to detect and treat Helicobacter pylori infection; surveillance endoscopy performed every three to five years, although there is limited evidence supporting this practice.
Genitourinary, pancreaticobiliary and breast cancers: At this time, there is insufficient evidence to recommend deviating from the population-based screening guidelines developed for the average-risk population. Screening efforts should be tailored to the individual's family history of the specific cancers. At the very least, patients with LS should continue to undergo population-based breast cancer screening, including mammography by age 40 years.
Familial adenomatous polyposis (FAP) and attenuated FAP (AFAP)
Hallazgos sobre la poliposis adenomatosa familiar
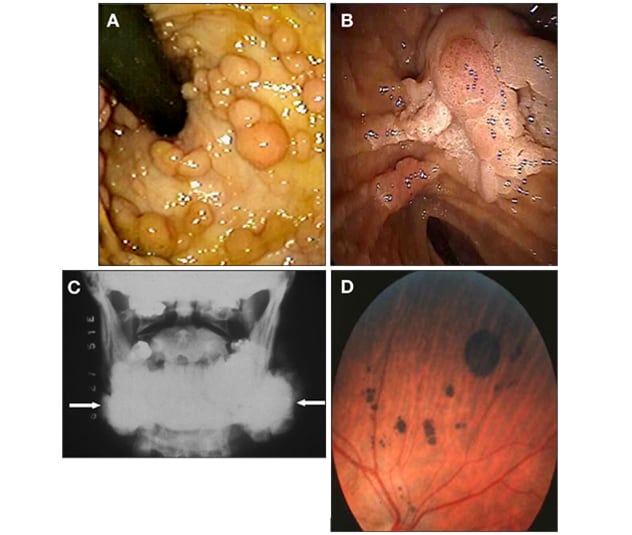
Hallazgos sobre la poliposis adenomatosa familiar
Hallazgos en pacientes con poliposis adenomatosa familiar. A. Paciente con numerosos pólipos en el recto y el colon. B. Vista endoscópica de la región periampular que muestra un gran pólipo adenomatoso en la ampolla y una extensión similar a una barba. C. Radiografía que muestra osteomas en la mandíbula. D. Fotografía del ojo que muestra abundante hipertrofia congénita de las lesiones del epitelio pigmentario de la retina.
Overview and clinical presentation
This rare, autosomal dominant, inherited condition presents with the development of hundreds of colorectal adenomas, generally during early adolescence. Although data about this syndrome are limited, the incidence of FAP is estimated to be 1 in 10,000 across all populations, and it accounts for less than 1% of all CRCs. In its early stages, FAP can present with nonspecific gastrointestinal symptoms (such as change in bowel movement pattern) and rectal bleeding.
"If left untreated, FAP is associated with a lifetime risk of CRC of nearly 100%," says Dr. Samadder. A less severe form of this disease, called AFAP, is characterized by later onset of polyps, fewer polyps (0 to 100 colon adenomas) and a lower overall risk of CRC (70%).
Genetic causes and diagnosis
Both FAP and AFAP are caused by germline mutations in the APC gene, a tumor suppressor gene that is part of the Wnt signaling pathway. Although FAP is an inherited disorder, up to 30% of cases of FAP are due to de novo APC mutations.
Genetic testing for FAP and AFAP should be considered in any of these situations:
- More than 10 adenomas detected during a single colonoscopy
- More than 20 adenomas in the colon or rectum over the patient's lifetime
- Known diagnosis of FAP in the family
- Family or personal history of early-onset CRC and extraintestinal features often associated with FAP, including congenital hypertrophy of the retinal pigment epithelium (CHRPE), osteomas and desmoid tumors
"FAP, AFAP and another hereditary cancer called MUTYH-associated polyposis have similar features, so confirming a molecular diagnosis of these syndromes via genetic testing is critical," says Dr. Samadder. Patients with classic FAP symptoms whose genetic testing results do not confirm an APC mutation may have other mutations.
Colorectal cancer surveillance
"Aggressive early diagnosis and management are warranted for patients with FAP," explains Dr. Samadder.
Recommendations: For FAP, colonoscopy is typically initiated by age 10 to 12 years and repeated annually, until the number of polyps present makes endoscopic management impractical. For AFAP, colonoscopy can begin at a slightly later age (late teens) and be repeated every one to two years.
Surgical management
Colectomy is generally recommended for patients with FAP and any of the following findings:
- Confirmed diagnosis of CRC
- Advanced histologic features in polyps (villous or high-grade dysplasia)
- Large adenomatous polyps (> 1 cm)
- Polyp burden exceeds capacity for endoscopic control (> 20 to 40 adenomatous polyps)
Proctocolectomy with ileal pouch-anal anastomosis and colectomy with ileorectal anastomosis are the two most commonly performed surgical approaches for patients with FAP. "The burden of disease in the rectum and the capacity to continue annual surveillance help guide the choice between these two surgical options," says Dr. Samadder.
Patients with an ileorectal anastomosis have a continued substantial risk of rectal cancer and require continued endoscopic surveillance every six to 12 months. Similarly, patients with an ileal pouch-anal anastomosis or ileostomy should undergo periodic endoscopic surveillance because of increased cancer risk related to the presence of preneoplastic lesions that can develop in the ileum, J pouch and anal transition zone.
Extracolonic cancer surveillance and management
A few of the more common types of extracolonic cancers associated with FAP and available recommendations for their surveillance and management are described below.
Duodenal cancer: This is the most common extracolonic malignancy and a major cause of mortality in patients who have undergone risk-reducing colectomy. Up to 80% of patients with FAP will develop duodenal adenomas, and the lifetime risk of duodenal cancer is 12%. The Spigelman classification score can guide endoscopic surveillance intervals. Recommendation: Baseline endoscopy, performed at 25 to 30 years of age or just prior to colectomy and repeated at intervals (from every one to five years) based on the patient's Spigelman score and stage.
Gastric polyps: Although gastric polyps are a frequent finding, the lifetime risk of gastric cancer is only 1%. Recommendation: Biopsy when observed during endoscopic surveillance.
Cancers of the thyroid: The lifetime risk of these cancers is 2%. Most occur in women, and some data suggest that Hispanic individuals are at increased risk. Recommendation: Annual thyroid clinical examination and consideration for thyroid ultrasonography.
Hepatoblastomas: These very rare liver tumors occur mostly in males, and children under the age of five years are at greatest risk. Recommendation: Alpha fetoprotein and liver ultrasonography can be considered every three to six months.
Dr. Samadder and colleagues at Mayo Clinic's multidisciplinary inherited cancers clinic are available to help identify high-risk patients and families, tailor approaches to cancer screening to detect cancers earlier, and formulate more-effective individualized treatment protocols for these patients.
For more information
Samadder NJ, et al. Hereditary cancer syndromes — A primer on diagnosis and management, Part 2: Gastrointestinal cancer syndromes. Mayo Clinic Proceedings. 2019;94:1099.