Jan. 17, 2020
Hypophosphatasia (HPP) is an inborn error of metabolism with a broad spectrum of clinical severity caused by loss of function mutations in the gene encoding tissue nonspecific alkaline phosphatase (TNSALP). To date, more than 400 distinct mutations in the TNSALP gene (ALPL) have been identified in patients with HPP, leading to highly variable clinical phenotypes. Peter J. Tebben, M.D., Pediatric and Adolescent Medicine, at Mayo Clinic in Rochester, Minnesota, explains: "The prevalence of severe forms is estimated to be 1 in 100,000, while mild forms are likely much more prevalent."
Clinical manifestations
HPP has been classified into five major categories, depending on the age at diagnosis. In general, the younger an individual is at the time of symptom onset, the more severe the disease. Disease severity can range from death in the perinatal period when untreated or be limited to dental problems or fractures in adulthood.
نقص الفوسفاتاز أثناء فترة الولادة
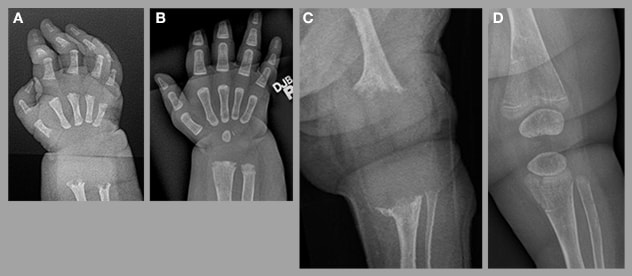
نقص الفوسفاتاز أثناء فترة الولادة
نقص الفوسفاتاز أثناء فترة الولادة، والذي يتسم بوجود نقص تمعدن حاد في الرسغ الأيمن (أ) والركبة اليسرى (ج)؛ تحسن كبير لاحق بعد تسعة أشهر من العلاج بدواء asfotase alfa (ب ود).
Perinatal HPP presents with clinical features noted either at birth or in utero based on prenatal ultrasound. Clinical exam reveals obvious skeletal abnormalities including chest wall deformities, as well as long bones that are short or bowed or both. The skeleton is hypomineralized. Historically, this form of HPP was frequently fatal. However, the availability of alkaline phosphatase enzyme replacement therapy (asfotase alfa) has significantly altered the natural history of perinatal HPP and most children survive.
Infantile HPP is diagnosed within the first six months of life. Characteristic changes of rickets are seen on radiographs, and fractures are often present. Infants fail to grow appropriately and some experience vitamin B6-responsive seizures. Hypercalcemia and nephrocalcinosis also are common. As with the perinatal form, morbidity and mortality are attenuated with asfotase alfa treatment.
Childhood HPP is diagnosed when disease manifests after 6 months of age. Children often have delays in gross motor milestones and static myopathy. A common feature of childhood HPP is premature loss of deciduous teeth (before 5 years of age) with the roots intact. Radiographs reveal changes consistent with rickets and often a radiolucent band extending from the growth plate into the metaphysis.
نقص الفوسفاتاز أثناء فترة البلوغ
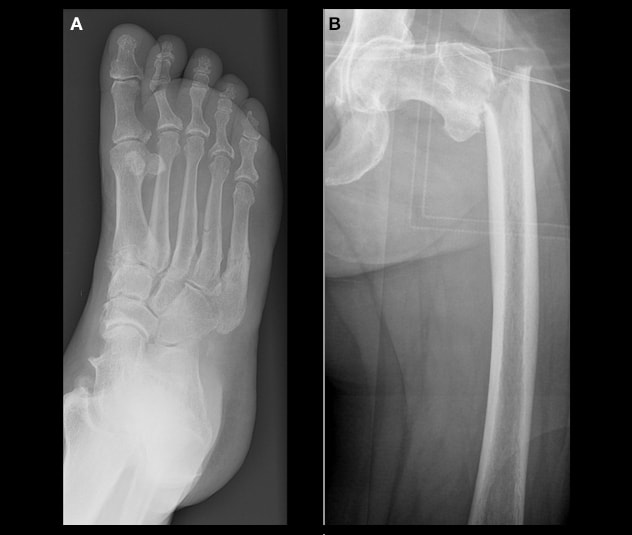
نقص الفوسفاتاز أثناء فترة البلوغ
نقص الفوسفاتاز في فترة البلوغ، والذي يتسم بوجود (أ) كسور في مشط القدم و(ب) كسر تحت المدور في الجانب الأيسر.
Adult HPP may manifest with recurrent or slow-to-heal metatarsal fractures or subtrochanteric femoral pseudofractures. A review of Mayo Clinic patients diagnosed with HPP as adults demonstrated that nonspecific musculoskeletal complaints were common at the time of presentation. Many adults with HPP report having had symptoms during childhood, despite formal diagnosis not being made until later in life.
Odontohypophosphatasia is the least severe form of HPP. Odontohypophosphatasia is diagnosed when dental abnormalities are present but no other skeletal disease, such as rickets or osteomalacia, is identified.
Laboratory findings
The hallmark laboratory finding in HPP is low alkaline phosphatase (ALP) activity. Because the abnormal ALPL gene located on chromosome 1 encodes the tissue nonspecific form of ALP (bone, liver, kidney), measuring bone-specific ALP is typically not necessary. Matthew T. Drake, M.D., Ph.D., Endocrinology, Diabetes, Metabolism and Nutrition, at Mayo Clinic in Rochester, Minnesota, cautions, "The presence of a low total ALP is not pathognomonic, as this finding can be associated with other disorders. Low total ALP may, however, indicate a diagnosis of HPP in the appropriate clinical context. In adults, prior treatment with anti-resorptive therapy is a common cause of low ALP levels."
When interpreting the result of an ALP measurement, it is crucial to use the appropriate age- and gender-specific reference values. Children have significantly higher ALP concentrations compared with adults. Accordingly, the diagnosis of HPP can be inadvertently overlooked if an adult reference range is applied.
Additional findings that are supportive of the diagnosis include hypercalcemia, hyperphosphatemia and hypercalciuria, particularly in the infantile and childhood forms. Urine phosphoethanolamine and serum pyridoxal 5'-phosphate are substrates for ALP and are elevated in patients with HPP. Pyridoxal 5'-phosphate is a product of vitamin B6, and patients taking supplements containing vitamin B6 should discontinue these supplements two weeks prior to testing.
Radiographic findings
Severely hypomineralized bone is seen in patients with the perinatal and infantile forms of the disease. Those with childhood HPP exhibit metaphyseal changes of rickets and bands of radiolucency extending from the growth plate into the metaphysis. In adults, metatarsal fractures, pseudofractures in the femur and calcium pyrophosphate deposition disease are commonly identified. Enlarged pulp chambers can be seen on dental X-rays.
Histologic findings
Bone histology varies depending on both the age of presentation and the severity of disease. Infantile HPP is characterized by severely defective skeletal mineralization, with osteoid composing the majority of bone tissue. On the other hand, patients with the childhood and adult forms of HPP with less severe manifestations may have no evidence of mineralization defects.
Iliac crest bone biopsies from patients with more-severe forms of childhood and adult HPP generally demonstrate osteomalacia, with areas of unmineralized osteoid and the absence of tetracycline label uptake. However, findings of osteomalacia can be patchy, with evidence of osteoid on some surfaces but comparatively normal mineralization on other bone surfaces.
Cortical and trabecular bone volumes are normal in adult HPP.
Genetic testing
HPP can be autosomal dominant or recessive, with most patients diagnosed using the clinical, biochemical and radiographic findings as described above. "In cases of prenatally identified HPP, genetic diagnosis is important to rule out other genetic etiologies of low bone mass," highlights David R. Deyle, M.D., Medical Genetics, at Mayo Clinic in Rochester, Minnesota. The diagnosis of HPP can be confirmed by the identification of either pathogenic biallelic variants of the gene encoding ALPL or a pathogenic heterozygous variant in ALPL.
Treatment
Asfotase alfa is approved for the treatment of children with HPP and for adults with childhood-onset disease. "Treatment has had a significant impact on the natural history of the more-severe perinatal, infantile and childhood forms of HPP," Dr. Tebben expands. "Treated infants can have dramatic improvement in skeletal mineralization, improved respiratory function and reduced mortality compared with historically untreated patients."
Patients treated with asfotase alfa for childhood HPP demonstrate improved motor capabilities. Much less information is available regarding asfotase alfa treatment of adults with HPP. As such, the treatment of adults with HPP remains an area in which more studies are needed to guide treatment decisions, particularly in less severely affected adults.
Dr. Drake concludes: "Recognition of adult HPP is important, as the most commonly utilized medications for osteoporosis (anti-resorptive agents) could further impair skeletal mineralization and should be avoided in patients with HPP."