Descripción general
La enfermedad de Huntington hace que las células nerviosas del cerebro se deterioren con el tiempo. La enfermedad afecta los movimientos, la capacidad de pensar y la salud mental de una persona.
La enfermedad de Huntington es poco frecuente. A menudo se transmite a través de una mutación genética del padre o la madre.
Los síntomas de la enfermedad de Huntington pueden presentarse en cualquier momento, pero a menudo empiezan cuando las personas tienen entre 30 y 40 años. Si la afección se presenta antes de los 20 años, recibe el nombre de enfermedad de Huntington juvenil. Cuando la enfermedad de Huntington se presenta de manera temprana, los síntomas pueden ser diferentes y es posible que la enfermedad progrese más rápido.
Hay medicamentos disponibles para ayudar a controlar los síntomas de la enfermedad de Huntington. Sin embargo, los tratamientos no pueden evitar el deterioro físico, mental y conductual causado por la enfermedad.
Productos y servicios
Síntomas
La enfermedad de Huntington suele causar trastornos motrices. También causa enfermedades mentales y problemas para pensar y planificar. Estas afecciones pueden causar una amplia gama de síntomas. Los primeros síntomas varían mucho de una persona a otra. Algunos síntomas parecen empeorar o afectar más a la capacidad funcional. Estos síntomas pueden variar en gravedad durante el transcurso de la enfermedad.
Trastornos del movimiento
Los trastornos motrices relacionados con la enfermedad de Huntington pueden causar corea, que son movimientos que no se pueden controlar. La corea son movimientos involuntarios que afectan a todos los músculos del cuerpo, sobre todo a los brazos y las piernas, la cara y la lengua. También puede afectar la capacidad de realizar movimientos voluntarios. Los síntomas pueden incluir los siguientes:
- Movimientos espasmódicos o de contorsión involuntarios
- Rigidez muscular o contracturas musculares
- Movimientos oculares lentos o inusuales
- Dificultad para caminar o mantener la postura y el equilibrio
- Dificultad para hablar o tragar
Las personas con enfermedad de Huntington tampoco pueden controlar los movimientos voluntarios. Esto puede tener un mayor impacto que los movimientos involuntarios causados por la enfermedad. Tener problemas con los movimientos voluntarios puede afectar la capacidad de una persona para trabajar, realizar actividades cotidianas, comunicarse y ser independiente.
Afecciones cognitivas
La enfermedad de Huntington suele causar problemas con las capacidades cognitivas. Entre estos síntomas pueden estar los siguientes:
- Dificultad para organizarse, establecer prioridades o enfocarse en tareas
- Perseveración, que es la falta de flexibilidad o quedarse atascado en un pensamiento, una conducta o una acción
- Falta de control de los impulsos, que puede tener como consecuencia arrebatos, actuar sin pensar y promiscuidad sexual
- Falta de conciencia sobre las conductas y aptitudes propias
- Lentitud para procesar pensamientos o ''encontrar'' las palabras indicadas
- Problemas para aprender información nueva
Afecciones de salud mental
La enfermedad mental más común asociada a la enfermedad de Huntington es la depresión. Y no se trata solamente de una reacción al recibir el diagnóstico de enfermedad de Huntington. Por el contrario, la depresión parece ocurrir debido a lesiones en el cerebro y cambios en el funcionamiento cerebral. Los síntomas pueden incluir los siguientes:
- Irritabilidad, tristeza o apatía
- Aislamiento social
- Problemas para dormir
- Fatiga y pérdida de energía
- Ideas sobre la muerte, morir o el suicidio
Otras enfermedades mentales comunes incluyen las siguientes:
- Trastorno obsesivo compulsivo, una afección caracterizada por pensamientos intrusivos que vuelven una y otra vez, y por comportamientos que se repiten una y otra vez
- Manía, que puede causar un estado de ánimo elevado, hiperactividad, conductas impulsivas y autoestima excesiva
- Trastorno bipolar, una afección con episodios alternados de depresión y manía
La pérdida de peso también es común en las personas con enfermedad de Huntington, sobre todo a medida que la enfermedad empeora.
Síntomas de la enfermedad de Huntington juvenil
En las personas más jóvenes, la enfermedad de Huntington comienza y evoluciona de manera un poco diferente a como lo hace en los adultos. Entre los síntomas que pueden aparecer al principio de la enfermedad, están los siguientes:
Cambios en la conducta
- Problemas para prestar atención
- Disminución repentina del rendimiento escolar general
- Problemas de comportamiento, como ser agresivo o perturbador
Cambios físicos
- Músculos contraídos y rígidos que afectan la marcha, especialmente en los niños pequeños
- Temblores, que son movimientos leves que no se pueden controlar
- Caídas frecuentes o torpeza
- Convulsiones
Cuándo consultar al médico
Consulta con el profesional de atención médica si observas cambios en tus movimientos, estado emocional o capacidad mental. Los síntomas de la enfermedad de Huntington pueden deberse a varias afecciones diferentes. Por lo tanto, es importante obtener un diagnóstico completo y rápido.
Causas
Patrón de la herencia autosómica dominante
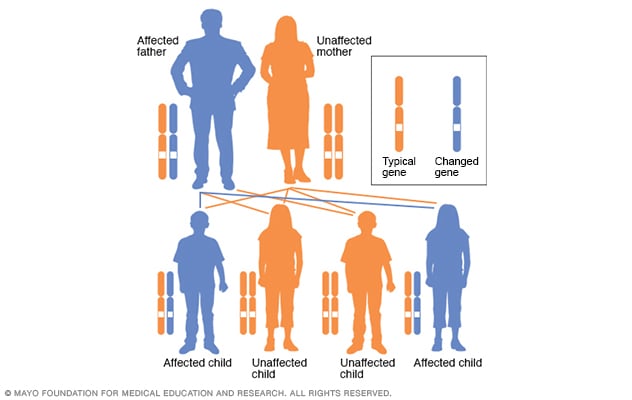
Patrón de la herencia autosómica dominante
En un trastorno autosómico dominante, el gen alterado es un gen dominante. Está ubicado en uno de los autosomas, que son los cromosomas no sexuales. Solo se necesita un gen alterado para que una persona se vea afectada por este tipo de afección. Una persona con una afección autosómica dominante, en este caso, el padre, tiene un 50 % de probabilidades de tener un hijo con un gen alterado y un 50 % de probabilidades de tener un hijo que no se vea afectado.
La enfermedad de Huntington es causada por una diferencia en un único gen que transmite el padre o la madre. La enfermedad de Huntington sigue un patrón autosómico dominante hereditario. Esto significa que una persona solo necesita una copia del gen atípico para desarrollar el trastorno.
A excepción de los cromosomas sexuales, una persona hereda dos copias de cada gen, una copia de cada progenitor. El padre o la madre con un gen atípico podría trasmitir la copia atípica del gen o la copia sana. Por ello, cada hijo e hija en la familia tiene una probabilidad del 50 % de heredar el gen que causa la afección genética.
Factores de riesgo
Las personas cuyo padre o madre tiene la enfermedad de Huntington corren el riesgo de presentar la enfermedad. Los hijos e hijas de un padre o una madre con Huntington tienen una probabilidad del 50 % de portar la mutación genética que causa la enfermedad.
Complicaciones
Tras el inicio de la enfermedad de Huntington, las capacidades funcionales de una persona empeoran progresivamente con el tiempo. La rapidez con la que empeora la enfermedad y el tiempo que tarda varían. El tiempo desde la aparición de los primeros síntomas hasta la muerte, por lo general, es de 10 a 30 años. La enfermedad de Huntington juvenil generalmente ocasiona la muerte en un plazo de 10 a 15 años después de la aparición de los síntomas.
La depresión vinculada a la enfermedad de Huntington puede aumentar el riesgo de suicidio. Algunas investigaciones sugieren que el riesgo de suicidio es mayor antes del diagnóstico y también cuando la persona pierde independencia.
Eventualmente, una persona que tiene la enfermedad de Huntington requerirá ayuda con todas las actividades cotidianas y los cuidados médicos. En las etapas finales de la enfermedad, la persona probablemente quede postrada en una cama y sin poder hablar. Por lo general, una persona con la enfermedad de Huntington puede comprender lo que se dice y reconoce a sus amigos y familiares; sin embargo, otras personas no reconocerán a sus familiares.
Las causas comunes de muerte comprenden las siguientes:
- Neumonía u otras infecciones
- Lesiones relacionadas con caídas
- Complicaciones relacionadas con problemas para tragar
Prevención
A las personas que tienen antecedentes familiares conocidos de enfermedad de Huntington les preocupa saber si les transmitirán el gen de Huntington a sus hijos e hijas. Estas personas pueden considerar pruebas genéticas y opciones de planificación familiar.
Si el padre o la madre que está en riesgo considera que le hagan pruebas genéticas, puede ser útil programar una cita con un consejero genético. El consejero genético te explicará los posibles riesgos de un resultado positivo de la prueba, que indicaría que el padre o la madre pueden presentar la enfermedad. Además, las parejas pueden tener que tomar decisiones adicionales sobre si tener hijos o considerar alternativas. Pueden optar por las pruebas prenatales para detectar el gen o por la fertilización in vitro con esperma u óvulos de un donante.
Otra opción para las parejas es la fertilización in vitro y el diagnóstico genético preimplantacional. En este proceso, se extraen los óvulos de los ovarios y se los fecundan con el esperma del padre en un laboratorio. Se analiza la presencia del gen Huntington en los embriones. Solo se implantan en el útero de la madre los que dan negativo en las pruebas del gen Huntington.
Fecundación in vitro
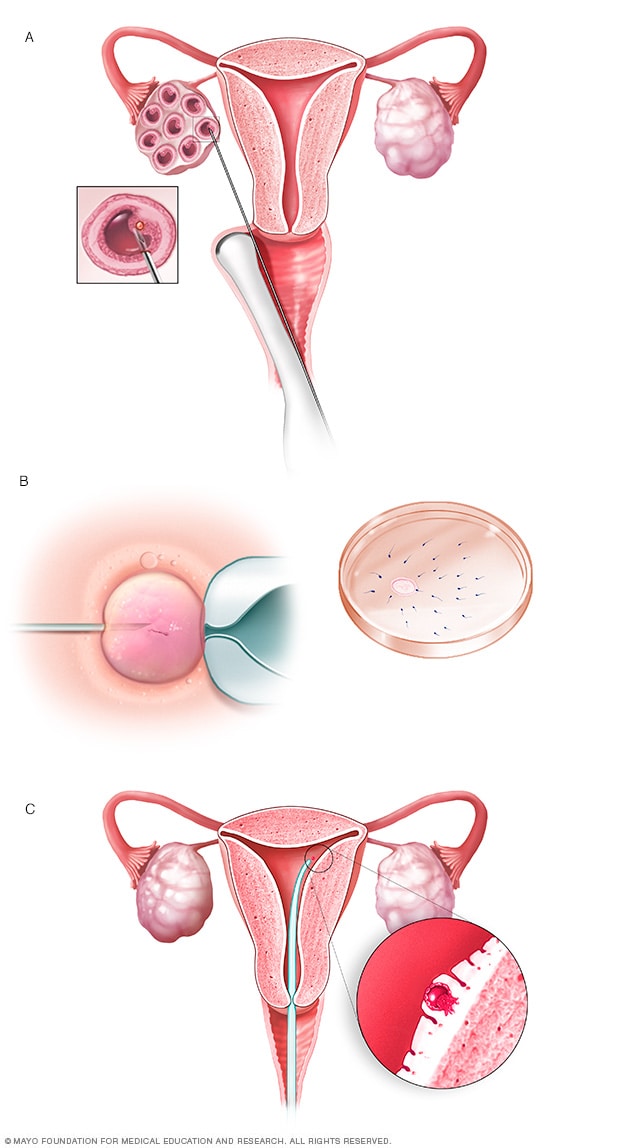
Fecundación in vitro
Durante la fertilización in vitro, los óvulos se extraen de unos sacos denominados folículos dentro de un ovario (A). Un óvulo se fecunda al inyectar un solo espermatozoide en el óvulo o al mezclar el óvulo con espermatozoides en una placa de Petri (B). El óvulo fecundado, o embrión, se transfiere al útero (C).
Aug. 20, 2024