Descripción general
El síndrome de QT largo es un trastorno de señalización cardíaca que puede hacer que el corazón lata rápido y de manera caótica (arritmias). Un trastorno de señalización cardíaca también se conoce como trastorno de conducción del corazón.
Algunas personas nacen con un ADN modificado que causa el síndrome de QT largo (síndrome de QT largo congénito). El síndrome de QT largo también puede aparecer más tarde en la vida (síndrome de QT largo adquirido) como consecuencia de algunas enfermedades, ciertos medicamentos o desequilibrios minerales.
El síndrome de QT largo puede provocar convulsiones o desmayos repentinos. Los jóvenes con el síndrome de QT largo presentan un mayor riesgo de muerte súbita.
El tratamiento para el síndrome de QT largo incluye cambios en el estilo de vida y medicamentos para prevenir latidos cardíacos peligrosos. A veces, se necesita cirugía para implantar un dispositivo con el fin de controlar el ritmo cardíaco.
Productos y servicios
Síntomas
Algunas personas con síndrome de QT largo no presentan ningún síntoma perceptible. Es posible que se descubra la afección cuando se haga un electrocardiograma o una prueba genética por otros motivos.
Los desmayos (síncope) son el síntoma más común del síndrome de QT largo. Es posible que los desmayos causados por el síndrome de QT largo ocurran con poca o ninguna advertencia. Algunas personas primero presentan signos de advertencia de desmayo, entre ellos:
- Visión borrosa
- Aturdimiento
- Latidos cardíacos fuertes (palpitaciones)
- Debilidad
El desmayo se produce cuando el corazón late temporalmente de forma desorganizada. Puedes desmayarte durante un momento de emoción, enojo, miedo o cuando haces ejercicio. Las cosas que te asustan, por ejemplo, un teléfono o un despertador, pueden hacer que pierdas el conocimiento.
El síndrome de QT largo también puede provocar convulsiones en algunas personas. En ocasiones, los síntomas del síndrome de QT largo pueden ocurrir durante el sueño.
La mayoría de las personas con síntomas del síndrome de QT largo tienen el primer episodio antes de los 40 años. Cuando la afección se presenta al nacer (síndrome de QT largo congénito), los síntomas pueden aparecer durante las primeras semanas a meses de vida o más adelante durante la infancia.
Por lo general, después de un episodio de QT largo, el corazón vuelve a su ritmo normal. Si el ritmo cardíaco no se restablece por sí solo o si no se emplea un desfibrilador externo a tiempo para restablecerlo, podría darse la muerte súbita.
Cuándo debes consultar con un médico
Llama al proveedor de atención médica si te desmayas repentinamente durante la actividad física o en un momento de mucha excitación, o después de haber tomado un medicamento nuevo. Si tienes un padre o madre, hermano o hijo con síndrome de QT largo, es importante que se lo digas al proveedor de atención médica. El síndrome de QT largo puede ser hereditario.
Causas
Cámaras y válvulas del corazón
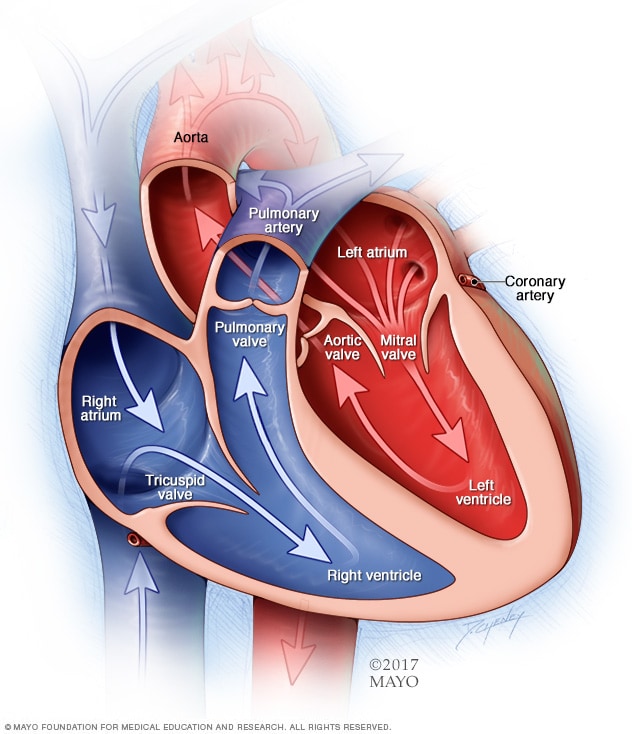
Cámaras y válvulas del corazón
Un corazón normal tiene dos cavidades superiores y dos cavidades inferiores. Las cavidades superiores, las aurículas derecha e izquierda, reciben la sangre entrante. Las cavidades inferiores, los ventrículos derecho e izquierdo más musculares, bombean la sangre desde el corazón hacia fuera. Las válvulas cardíacas, que mantienen el flujo sanguíneo en la dirección adecuada, son puertas en las aberturas de las cavidades.
El síndrome de QT largo es un trastorno del ritmo cardíaco cuya causa son los cambios en el sistema de recarga eléctrica del corazón. No afecta la estructura del corazón.
En un corazón normal, este envía sangre al cuerpo durante cada latido cardíaco. Para bombear la sangre, las cavidades del corazón se comprimen (contraen) y se relajan. Esta acción coordinada es controlada por el sistema eléctrico del corazón. Las señales eléctricas (impulsos) viajan desde la parte superior hasta la parte inferior del corazón. Estas señales le indican al corazón que se contraiga y lata. Después de cada latido cardíaco, el sistema se recarga para prepararse para el próximo.
En el síndrome de QT largo, el sistema eléctrico del corazón tarda más de lo usual en recargarse entre un latido y otro. Este retraso se denomina intervalo QT largo. Se puede observar en un electrocardiograma.
El síndrome de QT largo se suele agrupar en dos categorías principales según la causa.
- Si naces con la afección, se llama síndrome de QT largo congénito. Algunas formas del síndrome de QT largo son el resultado de un ADN modificado que se trasmite de padres a hijos (hereditario).
- Si lo causa una afección médica subyacente o un medicamento, se llama síndrome de QT largo adquirido. Este tipo de síndrome de QT largo suele ser reversible cuando se identifica y se trata la causa subyacente.
Causas del síndrome de QT largo congénito
Hasta ahora, se ha vinculado más de una docena de genes al síndrome de QT largo. Los investigadores han identificado cientos de alteraciones dentro de estos genes.
Hay dos formas de síndrome de QT largo congénito:
- Síndrome de Romano-Ward (forma autosómica dominante). Esta forma más común ocurre en personas que heredan solo una única variante genética de uno de los padres.
- Síndrome de Jervell y Lange-Nielsen (forma autosómica recesiva). Los episodios de esta forma de síndrome de QT largo poco frecuente suelen ocurrir a muy temprana edad y son más graves. En este síndrome, los niños reciben las variantes genéticas alteradas de ambos padres. Los niños nacen con el síndrome de QT largo y sordera.
Causas del síndrome de QT largo adquirido
Más de 100 medicamentos, muchos de ellos comunes, pueden causar intervalos QT prolongados en personas por lo demás sanas.
Si un medicamento provoca el síndrome de QT largo adquirido, la afección puede denominarse síndrome de QT largo inducido por medicamentos. Los medicamentos que pueden causar el síndrome de QT largo adquirido incluyen los siguientes:
- Determinados antibióticos, como eritromicina (Eryc, Erythrocin, otros), azitromicina (Zithromax) y otros
- Determinados medicamentos antimicóticos, utilizados para tratar infecciones por levaduras vaginales
- Diuréticos que provocan un desequilibrio de electrolitos (bajo nivel de potasio, más comúnmente)
- Medicamentos para el ritmo cardíaco (antiarrítmicos) que prolongan el intervalo QT
- Algunos medicamentos antidepresivos y antipsicóticos
- Algunos medicamentos para evitar las náuseas
Informa siempre a tu proveedor de atención médica sobre todos los medicamentos que tomas, incluso los medicamentos que compras sin receta médica.
Entre las afecciones médicas que pueden causar el síndrome de QT largo adquirido, se incluyen las siguientes:
- Temperatura corporal inferior a los 95 grados Fahrenheit (37 grados Celsius), una condición denominada hipotermia
- Bajo nivel de calcio (hipocalcemia)
- Bajo nivel de magnesio (hipomagnesemia)
- Bajo nivel de potasio (hipopotasiemia)
- Tumor no canceroso de la glándula suprarrenal (feocromocitoma)
- Accidente cerebrovascular o sangrado en el cerebro (intracraneal)
- Baja actividad de la tiroides (hipotiroidismo)
Factores de riesgo
Es posible que los siguientes factores incrementen el riesgo de tener síndrome de QT largo:
- Antecedentes de paro cardíaco
- Tener un padre, hermano o hijo con síndrome de QT largo
- Usar medicamentos que se sabe que causan intervalos QT prolongados
- Ser mujer y tomar medicamentos para el corazón
- Tener vómitos o diarrea excesivos, que provocan desequilibrios hidroelectrolíticos
- Tener trastornos alimentarios, como la anorexia nerviosa, que causan desequilibrios electrolíticos
Si tienes síndrome de QT largo y estás pensando en quedar embarazada, avisa a tu proveedor de atención médica. El proveedor de atención médica querrá controlarte de cerca durante el embarazo para prevenir cosas que puedan desencadenar un episodio de síndrome de QT largo.
Complicaciones
El tratamiento médico adecuado y los cambios en el estilo de vida pueden ayudar a prevenir las complicaciones relacionadas con el síndrome de QT largo.
Las complicaciones potenciales de este síndrome incluyen las siguientes:
-
Torsades de pointes (taquicardia ventricular en entorchado). Es un latido cardíaco irregular (arritmia) que puede poner en riesgo la vida. Las dos cavidades inferiores del corazón (ventrículos) laten rápido y de manera descontrolada, por lo que las ondas en el monitor de un electrocardiograma parecen torcidas. El corazón bombea menos sangre. La falta de sangre al cerebro causa un desmayo repentino y, a menudo, sin previo aviso.
Si el episodio dura mucho tiempo, el desmayo puede ir seguido de una convulsión de cuerpo entero. Si el peligroso ritmo no se corrige por sí solo, se produce una arritmia mortal llamada fibrilación ventricular.
- Fibrilación ventricular. Esta afección hace que las cavidades inferiores del corazón latan tan rápido que el corazón se estremece y deja de bombear sangre. A menos que se utilice un desfibrilador para que el corazón retome su ritmo normal, la fibrilación ventricular puede producir daño cerebral y muerte súbita.
- Muerte súbita. El síndrome de QT largo se ha asociado a la muerte súbita en personas jóvenes que parecen saludables. Esta afección podría ser la causa de algunos eventos en niños y adultos jóvenes, como desmayos, ahogos o convulsiones, sin motivo aparente.
Prevención
Los controles médicos regulares y una buena comunicación con el proveedor de atención médica pueden ayudar a prevenir afecciones médicas que conducen a algunos tipos de síndrome de QT largo adquirido. En especial, es importante evitar los medicamentos que pueden afectar el ritmo cardíaco y causar un intervalo QT largo.
Se desconoce cómo se puede prevenir el síndrome de QT largo congénito. Las familias con síndrome de QT largo hereditario podrían considerar la posibilidad de someterse a exámenes de detección genética. Con el tratamiento adecuado, puedes tratar y prevenir los latidos cardíacos peligrosos que pueden conducir a complicaciones del síndrome de QT largo.
Jan. 12, 2024