Descripción general
La fenilcetonuria es un trastorno hereditario poco frecuente que provoca que un aminoácido denominado fenilalanina se acumule en el cuerpo. La fenilcetonuria se produce como consecuencia de un cambio en el gen de la fenilalanina hidroxilasa (PAH, por sus siglas en inglés). Este gen ayuda a crear la enzima necesaria para descomponer la fenilalanina.
Sin la enzima necesaria para descomponer la fenilalanina, se puede desarrollar una peligrosa acumulación cuando una persona con fenilcetonuria ingiere alimentos que contienen proteína o consume aspartamo, un edulcorante artificial. Esto puede provocar, con el tiempo, graves problemas de salud.
Durante el resto de sus vidas, las personas con fenilcetonuria (bebés, niños y adultos) deben seguir una dieta que limite la fenilalanina, que se encuentra mayormente en alimentos con proteínas. Los medicamentos más novedosos pueden permitir que algunas personas con fenilcetonuria lleven una alimentación con una cantidad ilimitada o mayor cantidad de fenilalanina.
En los Estados Unidos y muchos otros países, se examina a los bebés poco después del nacimiento para detectar si tienen fenilcetonuria. Si bien no existe una cura para la fenilcetonuria, reconocerla y comenzar de inmediato un tratamiento puede ayudar a prevenir limitaciones en las áreas del pensamiento, la comprensión y la comunicación (discapacidad intelectual) y problemas de salud más graves.
Productos y servicios
Síntomas
Los recién nacidos que tienen fenilcetonuria al principio no presentan ningún síntoma. Sin embargo, sin tratamiento, los bebés suelen manifestar signos de fenilcetonuria en pocos meses.
Los signos y síntomas de la fenilcetonuria que no se trata pueden ser leves o graves, e incluyen los siguientes:
- Olor similar al de la humedad en el aliento, la piel o la orina, provocado por demasiada fenilalanina en el cuerpo.
- Problemas del sistema nervioso (neurológicos) que pueden incluir convulsiones.
- Erupciones cutáneas, como eczema.
- Piel, cabello y ojos más claros que los de los miembros de la familia, porque la fenilalanina no puede transformarse en melanina (pigmento responsable del color de la piel y del cabello).
- Tamaño de la cabeza inusualmente pequeño (microcefalia).
- Hiperactividad.
- Discapacidad intelectual.
- Retraso en el desarrollo.
- Problemas de comportamiento, emocionales y sociales.
- Trastornos de salud mental.
La gravedad varía
La gravedad de la fenilcetonuria depende del tipo.
- Fenilcetonuria clásica. La forma más grave del trastorno se llama fenilcetonuria clásica. Lo que sucede es que falta la enzima que se necesita para descomponer la fenilalanina, o bien hay una reducción grave de esta. Como consecuencia, los niveles elevados de fenilalanina pueden generar un daño cerebral grave.
- Formas menos graves de fenilcetonuria. En formas leves o moderadas, la enzima todavía tiene un poco de función, por lo que los niveles de fenilalanina no son tan elevados, lo que se traduce en un menor riesgo de tener daño cerebral significativo.
Independientemente de la forma, la mayoría de los bebés, niños y adultos que tienen este trastorno requieren una dieta especial para la fenilcetonuria a fin de prevenir la discapacidad intelectual y otras complicaciones.
Embarazo y fenilcetonuria
Las mujeres que tienen fenilcetonuria y quedan embarazadas corren el riesgo de tener otra forma de la enfermedad llamada fenilcetonuria materna. Si las mujeres no siguen la dieta especial para la fenilcetonuria antes del embarazo y durante este, los niveles de fenilalanina en la sangre pueden llegar a ser altos y dañar el bebé en desarrollo.
Incluso las mujeres que tienen formas más leves de esta afección pueden poner en riesgo al feto si no siguen la dieta para la fenilcetonuria.
Los bebés nacidos de mujeres que tienen niveles altos de fenilalanina no suelen heredar la fenilcetonuria. Sin embargo, pueden tener problemas graves si el nivel de fenilalanina en la sangre de la madre es alto durante el embarazo. Cuando nace, el bebé puede tener lo siguiente:
- Bajo peso al nacer
- Cabeza inusualmente pequeña
- Problemas en el corazón
Además, la fenilcetonuria materna puede hacer que el bebé tenga desarrollo tardío, discapacidad intelectual y problemas del comportamiento.
Cuándo debes consultar a un médico
Habla con tu proveedor de atención médica ante estas situaciones:
- Recién nacidos. Si el cribado neonatal de rutina demuestra que tu bebé podría tener fenilcetonuria, el proveedor de atención médica del niño iniciará un tratamiento alimentario de inmediato para prevenir problemas a largo plazo.
- Mujeres en edad fértil. Es especialmente importante para las mujeres que tienen antecedentes de fenilcetonuria que consulten con un proveedor de atención médica y que sigan una dieta para la fenilcetonuria antes de quedar embarazadas y durante el embarazo. Esto disminuye el riesgo de que los niveles altos de fenilalanina en la sangre hagan daño al feto.
- Adultos. Las personas que tienen fenilcetonuria necesitan atención médica de por vida. Es posible que los adultos que tienen fenilcetonuria y que suspendieron la dieta para esta enfermedad en la adolescencia puedan beneficiarse de una consulta con el proveedor de atención médica. Retomar la dieta puede mejorar el funcionamiento mental y el comportamiento, así como prevenir el mayor daño al sistema nervioso central que pueden provocar los niveles altos de fenilalanina.
Causas
Patrón hereditario autosómico recesivo
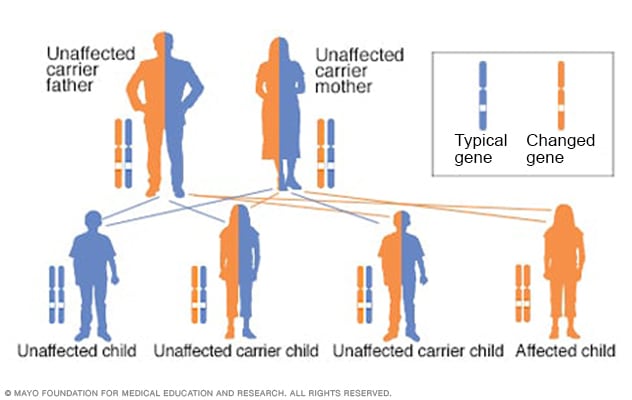
Patrón hereditario autosómico recesivo
Al tener un trastorno autosómico recesivo, heredas dos genes alterados, que a veces se denominan mutaciones. Recibes uno de cada progenitor. La salud de los padres con poca frecuencia se ve afectada porque tienen un solo gen alterado. Dos personas portadoras tienen un 25 % de probabilidades de tener un hijo no afectado, con dos genes no afectados. Tienen un 50 % de probabilidades de tener un hijo no afectado que también sea portador. Tienen un 25 % de probabilidades de tener un hijo afectado, con dos genes alterados.
Un cambio en los genes (mutación genética) provoca fenilcetonuria, que puede ser leve, moderada o grave. En una persona que tiene fenilcetonuria, un cambio en el gen de la fenilalanina hidroxilasa provoca la falta o una cantidad reducida de las enzimas necesarias para procesar la fenilalanina, un aminoácido.
Se puede presentar una acumulación peligrosa de fenilalanina cuando una persona con fenilcetonuria consume alimentos con un alto contenido de proteínas, como leche, queso, nueces o carne, granos, como pan y pasta, o aspartamo, un edulcorante artificial.
Herencia
Para que un niño herede la fenilcetonuria, tanto la madre como el padre deben tener el gen alterado y trasmitirlo. Este patrón de herencia se denomina autosómico recesivo.
Es posible que uno de los padres sea portador, es decir, que tenga el gen alterado que causa la fenilcetonuria, pero que no tenga la enfermedad. Si solo uno de los padres tiene el gen alterado, no hay riesgo de trasmitirle la fenilcetonuria al hijo, pero es posible que el niño sea portador.
Es más frecuente que dos padres que son portadores de este gen alterado sin saberlo les trasmitan la fenilcetonuria a sus hijos.
Factores de riesgo
Los siguientes son algunos factores de riesgo de heredar fenilcetonuria:
- Tener ambos padres con una modificación genética que cause fenilcetonuria. Los dos padres deben trasmitir una copia del gen modificado para que su hijo sufra esta afección.
- Ser descendiente de determinada raza o etnia. La fenilcetonuria afecta a personas de la mayoría de las etnias del mundo. Sin embargo, en los Estados Unidos es más común en las personas descendientes de europeos y mucho menos común en las personas afrodescendientes.
Complicaciones
La fenilcetonuria no tratada puede causar complicaciones en los bebés, los niños y los adultos que padecen el trastorno. Si las mujeres con esta afección tienen niveles elevados de fenilalanina en la sangre durante el embarazo, esto puede dañar al bebé no nacido.
La fenilcetonuria no tratada puede provocar lo siguiente:
- Daño cerebral irreversible y discapacidad intelectual marcada desde los primeros meses de vida
- Problemas neurológicos, como convulsiones y temblores
- En el caso de niños más grandes y adultos, problemas de conducta, emocionales y sociales
- Problemas graves de salud y desarrollo
Prevención
Si tienes síntomas de fenilcetonuria y piensas quedar embarazada:
- Sigue una dieta baja en fenilalanina. Las mujeres con fenilcetonuria pueden evitar dañar a su bebé en desarrollo si se ajustan o vuelven a una dieta baja en fenilalanina antes de quedar embarazadas. Los suplementos nutricionales diseñados para las personas con fenilcetonuria pueden garantizar una cantidad suficiente de proteínas y otros nutrientes durante el embarazo. Si tienes fenilcetonuria, habla con el proveedor de atención médica antes de buscar quedar embarazada.
- Considera la consejería genética. Si tienes fenilcetonuria, un pariente cercano con fenilcetonuria o un hijo que tiene fenilcetonuria, es posible que te beneficies de la consejería genética antes de la concepción. Un médico que se especializa en genética médica (genetista) puede ayudarte a comprender mejor cómo se trasmite la fenilcetonuria en la familia. El especialista también puede ayudarte a determinar el riesgo de tener un hijo con fenilcetonuria y asistirte con la planificación familiar.