Overview
What is cystic fibrosis? A Mayo Clinic expert explains
Learn more from pulmonologist Sarah Chalmers, M.D.
Hello. I'm Dr. Sarah Chalmers, a pulmonologist at Mayo Clinic. In this video, we'll cover the basics of cystic fibrosis. What is it? Who gets it? The symptoms, diagnosis and treatment. Whether you're looking for answers for yourself or someone you love, we're here to give you the best information available. Cystic fibrosis is a disorder that damages your lungs, digestive tract and other organs. It's an inherited disease caused by a defective gene that can be passed from generation to generation. Cystic fibrosis affects the cells that produce mucus, sweat and digestive juices. These secreted fluids are normally thin and slippery. But in people with CF, they're thick and sticky. Instead of acting as lubricants, these secretions plug up the tubes, ducts and airways in your body. Although there is no cure for cystic fibrosis, people with this condition are generally able to live normal lives. There are many tools and techniques doctors use to help manage this complicated condition and with improvement in screening and treatments, life expectancy for those with cystic fibrosis is better than ever before.
Simply put, cystic fibrosis is a gene defect. A defect to this gene changes how a salt moves in and out of cells, resulting in thick, sticky mucus in the respiratory, digestive and reproductive systems. It's an inherited condition. A child needs to inherit one copy of the mutated gene from each parent to develop cystic fibrosis. If they only inherit one copy from one parent, they won't develop it. However, they will be a carrier of that mutated gene, so they could pass it along to their own children in the future. Because CF is an inherited disorder, family history determines your risk. Although it can occur in all races, cystic fibrosis is most common in white people of North European ancestry.
There are two kinds of symptoms associated with cystic fibrosis. The first are respiratory symptoms. Thick, sticky mucus can clog the tubes that carry air in and out of your lungs. This can trigger a persistent cough that produces thick mucus, wheezing, exercise intolerance, repeated lung infections, and inflamed nasal passages or a stuffy nose or recurrent sinusitis. The second type of symptoms are digestive. That same thick mucus that can clog your airways can also bog tubes that carry enzymes from your pancreas to your small intestine. This can result in foul-smelling or greasy stools, poor weight gain and growth, intestinal blockage, or chronic and severe constipation, which may include frequent straining while trying to pass stool. If you or your child show symptoms of cystic fibrosis or if someone in your family has CF, talk with your doctor about testing for the disease.
Since this disease is an inherited condition, reviewing your family history is important. Genetic testing may be done to see if you carry the mutated gene that triggers cystic fibrosis. A sweat test may also be conducted. CF causes higher than normal levels of salt in your sweat. Doctors will examine the levels of salt in your sweat to confirm a diagnosis.
Because this condition is passed from parent to children, newborn screening is routinely done in every state in the U.S. Early diagnosis of CF means that treatments can begin immediately. Unfortunately, there is no cure for cystic fibrosis, but proper treatment can ease your symptoms, reduce complications, and improve your quality of life. Doctors may decide that certain medications are necessary. These could include antibiotics to treat and prevent lung infections, anti-inflammatories to lessen the swelling in your airways, or mucus-thinning drugs to help expel mucus and improve lung function. Medications can also help improve digestive function. From stool softeners to enzymes, to acid-reducing drugs. Some medications can even target the gene defect that causes cystic fibrosis, aiding the faulty proteins to improve lung function and reduce salt in your sweat. Outside of medications, airway clearance techniques, also called chest physical therapy, can relieve mucus obstruction and help to reduce infection and inflammation in the airways. These techniques loosen the thick mucus in the lungs, making it easier to cough up. In some cases, doctors turn to surgery to help alleviate conditions that can arise from cystic fibrosis. For instance, nasal and sinus surgery to help you breathe, or bowel surgery to help improve digestive function. In life-threatening instances, lung transplant and liver transplant had been performed. Managing cystic fibrosis can be very complex. So consider getting treatment at a center with medical professionals trained in the disorder to evaluate and treat your condition. You can even ask your physician about clinical trials. New treatments, interventions and tests are constantly under development to help prevent, detect, and treat this disease.
Learning you or someone you know has cystic fibrosis can be incredibly challenging. It's okay to feel depressed, anxious, angry, or afraid. In time, you'll find ways to cope, find support and talk to others who are going through it too. Look to your friends and family to help manage stress and reduce anxiety. Seek professional help. Remember, physical conditions come with an emotional and mental burden. And take the time to learn about cystic fibrosis. It's a complicated, severe disorder. So don't hesitate to talk to your medical team about your questions or concerns. With the knowledge and treatment available to doctors today, life with cystic fibrosis is better than ever before. If you'd like to learn even more about cystic fibrosis, watch our other related videos or visit mayoclinic.org. We wish you well.
Cystic fibrosis
Cystic fibrosis
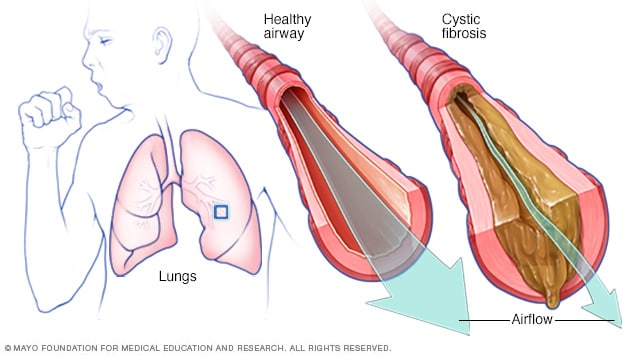
In cystic fibrosis, the airways fill with thick, sticky mucus, making it difficult to breathe. The thick mucus is also an ideal breeding ground for bacteria and fungi.
Cystic fibrosis (CF) is an inherited disorder that causes severe damage to the lungs, digestive system and other organs in the body.
Cystic fibrosis affects the cells that produce mucus, sweat and digestive juices. These secreted fluids are normally thin and slippery. But in people with CF, a defective gene causes the secretions to become sticky and thick. Instead of acting as lubricants, the secretions plug up tubes, ducts and passageways, especially in the lungs and pancreas.
Although cystic fibrosis is progressive and requires daily care, people with CF are usually able to attend school and work. They often have a better quality of life than people with CF had in previous decades. Improvements in screening and treatments mean that people with CF now may live into their mid- to late 30s or 40s, and some are living into their 50s.
Products & Services
Symptoms
In the U.S., because of newborn screening, cystic fibrosis can be diagnosed within the first month of life, before symptoms develop. But people born before newborn screening became available may not be diagnosed until the signs and symptoms of CF show up.
Cystic fibrosis signs and symptoms vary, depending on the severity of the disease. Even in the same person, symptoms may worsen or improve as time passes. Some people may not experience symptoms until their teenage years or adulthood. People who are not diagnosed until adulthood usually have milder disease and are more likely to have atypical symptoms, such as recurring bouts of an inflamed pancreas (pancreatitis), infertility and recurring pneumonia.
People with cystic fibrosis have a higher than normal level of salt in their sweat. Parents often can taste the salt when they kiss their children. Most of the other signs and symptoms of CF affect the respiratory system and digestive system.
Respiratory signs and symptoms
The thick and sticky mucus associated with cystic fibrosis clogs the tubes that carry air in and out of your lungs. This can cause signs and symptoms such as:
- A persistent cough that produces thick mucus (sputum)
- Wheezing
- Exercise intolerance
- Repeated lung infections
- Inflamed nasal passages or a stuffy nose
- Recurrent sinusitis
Digestive signs and symptoms
The thick mucus can also block tubes that carry digestive enzymes from your pancreas to your small intestine. Without these digestive enzymes, your intestines aren't able to completely absorb the nutrients in the food you eat. The result is often:
- Foul-smelling, greasy stools
- Poor weight gain and growth
- Intestinal blockage, particularly in newborns (meconium ileus)
- Chronic or severe constipation, which may include frequent straining while trying to pass stool, eventually causing part of the rectum to protrude outside the anus (rectal prolapse)
When to see a doctor
If you or your child has symptoms of cystic fibrosis — or if someone in your family has CF — talk with your doctor about testing for the disease. Consult a physician who is knowledgeable about CF.
Cystic fibrosis requires consistent, regular follow-up with your doctor, at least every three months. Contact you doctor if you experience new or worsening symptoms, such as more mucus than usual or a change in the mucus color, lack of energy, weight loss, or severe constipation.
Seek immediate medical care if you're coughing up blood, have chest pain or difficulty breathing, or have severe stomach pain and distention.
Causes
In cystic fibrosis, a defect (mutation) in a gene — the cystic fibrosis transmembrane conductance regulator (CFTR) gene — changes a protein that regulates the movement of salt in and out of cells. The result is thick, sticky mucus in the respiratory, digestive and reproductive systems, as well as increased salt in sweat.
Many different defects can occur in the gene. The type of gene mutation is associated with the severity of the condition.
Children need to inherit one copy of the gene from each parent in order to have the disease. If children inherit only one copy, they won't develop cystic fibrosis. However, they will be carriers and could pass the gene to their own children.
Risk factors
Because cystic fibrosis is an inherited disorder, it runs in families, so family history is a risk factor. Although CF occurs in all races, it's most common in white people of Northern European ancestry.
Complications
Complications of cystic fibrosis can affect the respiratory, digestive and reproductive systems, as well as other organs.
Respiratory system complications
- Damaged airways (bronchiectasis). Cystic fibrosis is one of the leading causes of bronchiectasis, a chronic lung condition with abnormal widening and scarring of the airways (bronchial tubes). This makes it harder to move air in and out of the lungs and clear mucus from the bronchial tubes.
- Chronic infections. Thick mucus in the lungs and sinuses provides an ideal breeding ground for bacteria and fungi. People with cystic fibrosis may often have sinus infections, bronchitis or pneumonia. Infection with bacteria that is resistant to antibiotics and difficult to treat is common.
- Growths in the nose (nasal polyps). Because the lining inside the nose is inflamed and swollen, it can develop soft, fleshy growths (polyps).
- Coughing up blood (hemoptysis). Bronchiectasis can occur next to blood vessels in the lungs. The combination of airway damage and infection can result in coughing up blood. Often this is only a small amount of blood, but it can also be life-threatening.
- Pneumothorax. In this condition, air leaks into the space that separates the lungs from the chest wall, and part or all of a lung collapses. This is more common in adults with cystic fibrosis. Pneumothorax can cause sudden chest pain and breathlessness. People often feel a bubbling sensation in the chest.
- Respiratory failure. Over time, cystic fibrosis can damage lung tissue so badly that it no longer works. Lung function usually worsens gradually, and it eventually can become life-threatening. Respiratory failure is the most common cause of death.
- Acute exacerbations. People with cystic fibrosis may experience worsening of their respiratory symptoms, such as coughing with more mucus and shortness of breath. This is called an acute exacerbation and requires treatment with antibiotics. Sometimes treatment can be provided at home, but hospitalization may be needed. Decreased energy and weight loss also are common during exacerbations.
Digestive system complications
- Nutritional deficiencies. Thick mucus can block the tubes that carry digestive enzymes from your pancreas to your intestines. Without these enzymes, your body can't absorb protein, fats or fat-soluble vitamins, so you can't get enough nutrients. This can result in delayed growth, weight loss or inflammation of the pancreas.
- Diabetes. The pancreas produces insulin, which your body needs to use sugar. Cystic fibrosis increases the risk of diabetes. About 20% of teenagers and 40% to 50% of adults with CF develop diabetes.
- Liver disease. The tube that carries bile from your liver and gallbladder to your small intestine may become blocked and inflamed. This can lead to liver problems, such as jaundice, fatty liver disease and cirrhosis — and sometimes gallstones.
- Intestinal obstruction. Intestinal blockage can happen to people with cystic fibrosis at all ages. Intussusception, a condition in which a segment of the intestine slides inside an adjacent section of the intestine like a collapsible telescope, also can occur.
- Distal intestinal obstruction syndrome (DIOS). DIOS is partial or complete obstruction where the small intestine meets the large intestine. DIOS requires urgent treatment.
Reproductive system complications
- Infertility in men. Almost all men with cystic fibrosis are infertile because the tube that connects the testes and prostate gland (vas deferens) is either blocked with mucus or missing entirely. Certain fertility treatments and surgical procedures sometimes make it possible for men with CF to become biological fathers.
- Reduced fertility in women. Although women with cystic fibrosis may be less fertile than other women, it's possible for them to conceive and to have successful pregnancies. Still, pregnancy can worsen the signs and symptoms of CF, so be sure to discuss the possible risks with your doctor.
Other complications
- Thinning of the bones (osteoporosis). People with cystic fibrosis are at higher risk of developing a dangerous thinning of bones. They may also experience joint pain, arthritis and muscle pain.
- Electrolyte imbalances and dehydration. Because people with cystic fibrosis have saltier sweat, the balance of minerals in their blood may be upset. This makes them prone to dehydration, especially with exercise or in hot weather. Signs and symptoms include increased heart rate, fatigue, weakness and low blood pressure.
- Mental health problems. Dealing with a chronic illness that has no cure may cause fear, depression and anxiety.
Prevention
If you or your partner has close relatives with cystic fibrosis, you both may choose to have genetic testing before having children. The test, which is performed in a lab on a sample of blood, can help determine your risk of having a child with CF.
If you're already pregnant and the genetic test shows that your baby may be at risk of cystic fibrosis, your doctor can conduct additional tests on your developing child.
Genetic testing isn't for everyone. Before you decide to be tested, you should talk to a genetic counselor about the psychological impact the test results might carry.
Nov. 23, 2021